


Die Huntington-Krankheit ist eine Erbkrankheit, die mit unwillkürlichen, vereinzelt auftretenden ruck- oder krampfartigen Bewegungen beginnt und im Verlauf zu auffälligeren unwillkürlichen Bewegungen (Chorea und Athetose), einem geistigen Verfall und zum Tod führt.
Bei der Huntington-Krankheit werden Teile des Gehirns abgebaut, die für fließende Bewegungen sorgen und diese koordinieren.
Die Bewegungen werden ruckartig und unkoordiniert, und die geistige Funktion, einschließlich der Selbstkontrolle und des Gedächtnisses, verschlechtert sich.
Die Ärzte gründen die Diagnose auf Symptome, die Familienanamnese, Aufnahmen des Gehirns und Gentests.
Arzneimittel können zur Linderung der Symptome beitragen, doch die Erkrankung schreitet voran und endet letztendlich mit dem Tod.
(Siehe auch Überblick über Bewegungsstörungen.)
Die Huntington-Krankheit betrifft 1 bis 10 von 100.000 Menschen. Die Anzahl der Betroffenen variiert abhängig davon, in welcher Gegend der Welt sie leben. Beide Geschlechter sind gleichermaßen betroffen.
Das Gen für die Huntington-Krankheit ist dominant. Das bedeutet, dass die Erkrankung auch verursacht wird, wenn man nur eine Kopie des veränderten Gens von einem Elternteil geerbt hat. Daher beträgt die Wahrscheinlichkeit diese Erkrankung zu entwickeln bei Kindern einer Person, die an der Huntington-Krankheit leidet, 50 Prozent.
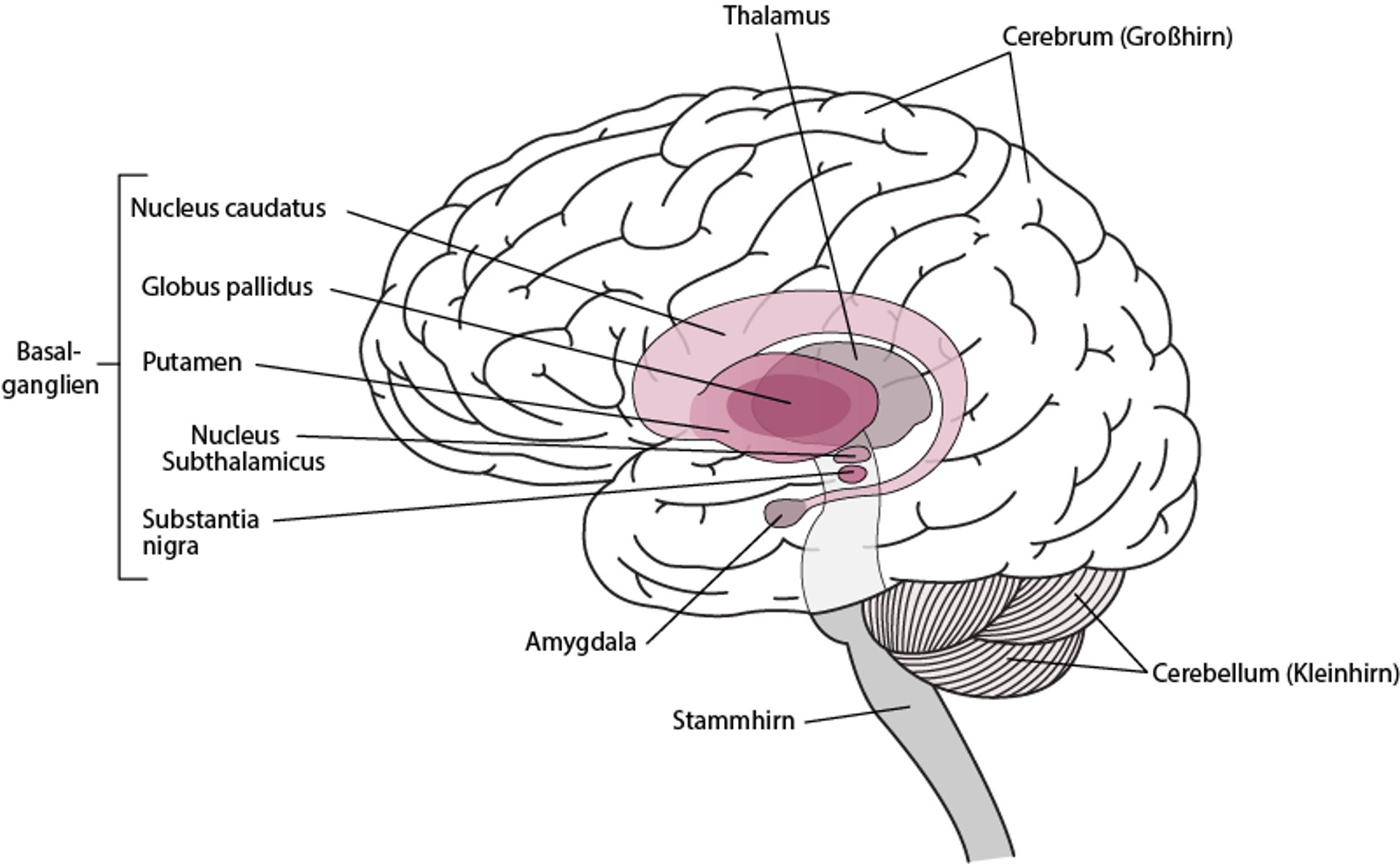
Die Chorea Huntington wird durch den allmählichen Abbau von Bereichen der Basalganglien, d. h. des Nucleus caudatus und des Putamen, verursacht. Die Basalganglien sind eine Ansammlung von Nervenzellen, die sich in der Hirnbasis, tief im Inneren des Gehirns, befinden. Sie helfen dabei, Bewegungen flüssig zu machen und zu koordinieren.
Lokalisation der Basalganglien
Die Basalganglien sind eine Ansammlung von Nervenzellen, die sich tief im Gehirn befinden. Hierzu zählen folgende:
Die Basalganglien helfen dabei, Muskelbewegungen auszulösen und geschmeidig zu machen, unwillkürliche Bewegungen zu unterdrücken und Veränderungen der Haltung zu koordinieren. |

Symptome der Huntington-Krankheit
Die Symptome der Huntington-Krankheit beginnen in der Regel unauffällig im Alter von 35 bis 40 Jahren, können sich jedoch auch vor dem Erwachsenenalter entwickeln.
In den frühen Stadien der Huntington-Krankheit können sich das Gesicht, der Rumpf und die Gliedmaßen unwillkürlich und schnell bewegen. Zuerst können die Betroffenen diese abnormalen unwillkürlichen Bewegungen in willkürliche Bewegungen umwandeln, so dass die abnormalen Bewegungen kaum auffallen. Im Laufe der Zeit werden die Bewegungen jedoch offensichtlicher.
Die Muskeln ziehen sich womöglich kurz und schnell zusammen, wodurch die Arme und andere Körperteile plötzlich zucken, manchmal mehrmals hintereinander.
Die Betroffenen laufen möglicherweise auf tänzelnde oder übertrieben beschwingte Weise, wie eine Marionette. Die Betroffenen schneiden Grimassen, schlenkern die Glieder und blinzeln häufiger. Die Bewegungen werden unkoordiniert und langsamer. Letztendlich ist der gesamte Körper betroffen, sodass Laufen, Stillsitzen, Essen, Sprechen, Schlucken und Ankleiden extrem erschwert werden.
Geistige Veränderungen treten häufig vor oder während der Entwicklung der abnormalen Bewegungen auf. Diese Veränderungen sind zunächst kaum merkbar. Die Betroffenen können mit der Zeit immer reizbarer, erregbarer und aufgeregter werden. Sie können das Interesse an ihren gewöhnlichen Aktivitäten verlieren. Sie können sehr impulsiv werden, ihre Beherrschung verlieren, niedergeschlagen sein oder sexuell ausschweifend werden.
Mit dem Fortschreiten der Huntington-Krankheit verhalten sie sich möglicherweise immer verantwortungsloser und streifen oft ziellos umher. Im Laufe der Jahre verlieren sie ihr Gedächtnis und ihre Fähigkeit zum rationalen Denken. Sie können schwere Depressionen entwickelt und versuchen, Suizid zu begehen. Sie können auch ängstlich werden oder eine Zwangsstörung entwickeln.
Im fortgeschrittenen Stadium ist die Demenz stark ausgeprägt und die Betroffenen sind bettlägerig. Eine Vollzeitpflege oder die Pflege in einem Pflegeheim ist erforderlich. Normalerweise tritt der Tod innerhalb von 13 bis 15 Jahren nach Symptombeginn ein.
Diagnose der Huntington-Krankheit
Eine ärztliche Untersuchung, bestätigt durch genetische Tests
Computertomografie oder Magnetresonanztomografie
Die Huntington-Krankheit kann in den Frühstadien oft schwer zu diagnostizieren sein, da die Symptome unauffällig sind. Die Krankheit kann aufgrund von Symptomen und der Familienanamnese vermutet werden. Die Ärzte sollten über Verwandte informiert werden, die geistige Störungen hatten oder bei denen neurologische (wie die Parkinson-Krankheit) oder psychiatrische Störungen (wie Schizophrenie) diagnostiziert wurden, da sie möglicherweise an einer nicht diagnostizierten Huntington-Krankheit litten.
Computertomografien (CT) oder Magnetresonanztomografien (MRT) werden durchgeführt, um zu prüfen, ob die Basalganglien und andere Bereiche des Gehirns degeneriert sind, was für die Erkrankung charakteristisch ist, und um andere Erkrankungen auszuschließen.
Genetische Untersuchungen werden durchgeführt, um die Diagnose zu bestätigen. Genetische Untersuchungen und Beratungen sind wichtig für Menschen, die die Erkrankung in der Familiengeschichte haben, jedoch keine Symptome aufweisen, da diese Personen wahrscheinlich Kinder bekommen werden, bevor die Symptome erscheinen. Bei solchen Personen sollte der genetischen Untersuchung eine Genetikberatung vorausgehen. Sie werden zu Zentren überweisen, die das Fachwissen dazu haben, um mit den komplexen ethischen und psychologischen Problemen umzugehen, die damit verbunden sind.
Behandlung der Huntington-Krankheit
Antipsychotika und andere Arzneimittel zur Linderung der Symptome
Sobald die Diagnose gestellt wurde, sollten Personen mit Huntington-Krankheit eine Vorausverfügung dazu festlegen, welche Art der medizinischen Versorgung sie an ihrem Lebensende wünschen (Patientenverfügung).
Für die Huntington-Krankheit gibt es kein Heilmittel. Bestimmte Arzneimittel, darunter Antipsychotika (z. B. Chlorpromazin, Haloperidol, Risperidon und Olanzapin) können dabei helfen, die Aufgeregtheit unter Kontrolle zu bekommen. Medikamente, welche die Dopaminmenge senken (wie Tetrabenazin, Deutetrabenazin und das Antihypertensivum Reserpin) können dabei helfen, die auffälligen Bewegungen zu stoppen (unterdrücken).
Bei einer Depression können Antidepressiva angewendet werden.
Ärzte bieten den Eltern oder Geschwistern von Patienten mit Huntington-Krankheit eine genetische Beratung und Gentests an. Eine genetische Beratung sollte vor Gentests angeboten werden, da die Folgen der Huntington-Krankheit so schwerwiegend sind. Eine Beratung ist besonders wichtig für Frauen im gebärfähigen Alter und Männer, die erwägen, Vater zu werden.
Weitere Informationen
Bei dem Folgenden handelt es sich um ein englischsprachiges Hilfsmittel, das nützlich sein kann. Bitte beachten Sie, dass das MANUAL nicht für den Inhalt dieser Quelle verantwortlich ist.
Genetics Home Reference: Huntington-Krankheit: Auf dieser Website ist die Huntington-Krankheit beschrieben und es wird erörtert, wodurch sie verursacht und wie sie vererbt wird. Außerdem enthält sie Links zur Diagnose und Behandlung.