Der Kiefer kann bei der Geburt fehlen, deformiert oder unvollständig entwickelt sein, oft in Verbindung mit anderen angeborenen Anomalien und Syndromen.
(Siehe auch Einführung in angeborene kraniofaziale und muskuloskelettale Störungen und Überblick über angeborene kraniofaziale Anomalien.)
Wenn andere Anomalien vorliegen, kann ein klinischer Genetiker dabei helfen, die Evaluation anzuleiten, da die Identifizierung des zugrunde liegenden Syndroms für die Prognose und Familienberatung wichtig ist. Eine klinisch-genetische Untersuchung wird jedoch auch in Fällen scheinbar isolierter kongenitaler Anomalien empfohlen.
Bei der Untersuchung von Patienten mit kraniofazialen Anomalien sollten eine Chromosomen-Mikroarray-Analyse, spezifische Gentests oder umfassendere Gen-Panel-Tests in Betracht gezogen werden. Wenn die Ergebnisse dieser Tests nicht diagnostisch sind, wurde eine vollständige Exom-Sequenzanalyse empfohlen.
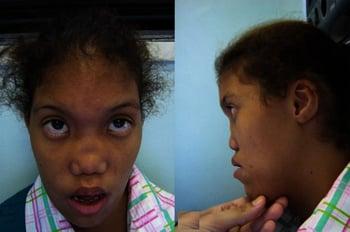
Mikrognathie (kleiner Unterkiefer)
Mikrognathie kann bei > 700 genetischen Syndromen auftreten.
Die Pierre-Robin-Sequenz ist eine häufige Manifestation der Mikrognathie, die durch einen U-förmigen Spalt des weichen Gaumens und durch eine Obstruktion der oberen Atemwege, die durch Glossoptose (einer Zunge, die an die Rückseite der Kehle zurückfällt) verursacht wird, gekennzeichnet ist; eine Schallleitungsschwerhörigkeit kann ebenfalls vorliegen. Das Füttern kann schwierig sein und manchmal kommt es zu zyanotischen Anfällen, weil die Zunge nach hinten fällt und den Pharynx verschließt. Eine aufrechte Haltung kann beim Füttern hilfreich sein. Das unkoordinierte Schlucken kann aber eine nasogastrale Sonde oder eine Gastrostomie notwendig machen. Wenn die Zyanose oder die Atemprobleme anhalten, muss manchmal eine Tracheotomie oder eine Operation, bei der die Zunge in einer vorderen Position (z. B. an der vorderen inneren Lippe) fixiert wird, durchgeführt werden. Eine otologische Untersuchung sollte veranlasst werden.
Etwa ein Drittel der Patienten mit Mikrognathie haben Anomalien, die auf eine zugrunde liegende Chromosomenstörung oder ein genetisches Syndrom hindeuten. Einige der Diagnosen, die berücksichtigt werden müssen, umfassen Treacher-Collins-Syndrom (mit schräg nach unten laufenden Augen, Kolobom des Augenlids, missgebildeter Ohrmuschel [Mikrotie] und Hörverlust assoziiert), Nager-Syndrom, Goldenhar- (oculoauriculovertebrales) Syndrom und zerebrocostomandibuläres Syndrom.
Die chirurgische Verlängerung des Unterkiefers kann Aussehen und Funktion verbessern. In dem üblichen Verfahren, der sogenannten Distraktionsosteogenese, wird eine Osteotomie durchgeführt, gefolgt von der Platzierung eines Teilungsgerät zwischen die beiden Teile. Mit der Zeit wird die Entfernung zwischen den zwei Teilen vergrößert und neuer Knochen wächst in dieser Lücke, um die Mandibula zu vergrößern.
Agnathie
Ein angeborenes Fehlen des kondyloiden Fortsatzes (manchmal auch des koronoiden Fortsatzes oder/und Teilen der Mandibula) stellt eine schwere Fehlbildung dar. Die Mandibula verlagert sich zur betroffenen Seite und führt zu einer schweren Malokklusion. Die nicht betroffene Seite ist verlängert und abgeflacht. Oft kommen auch gleichzeitig Fehlbildungen des äußeren, mittleren und inneren Ohres, des Temporalknochens, der Parotis, der Masseter und des Fazialisnervs vor. Syndrome, die berücksichtigt werden müssen, umfassen Agnathie-Holoprosenzephalie, Otozephalie, eine schwere Form des zerebrocostomandibulären Syndroms und Ivemark-Syndrom.
Die Röntgenbilder oder die Gesichts-CT der Mandibula und des temporomandibulären Gelenks zeigen das Ausmaß der Unterentwicklung und unterscheiden die Agenesie von anderen Krankheiten, die zu einer ähnlichen Gesichtfehlbildung führen, aber keine so schweren strukturellen Defekte mit sich bringen. Eine Gesichts-CT wird in der Regel vor der Operation durchgeführt.
Die Behandlung einer Agnathie besteht aus einer sofortigen Rekonstruktion mit autologem Knochenmaterial, um ein Fortschreiten der fazialen Fehlbildung zu begrenzen. Oft können eine Mentoplastik, darüberliegende Transplantate mit Knochen- und Knorpelmaterial, Weichteillappen und -transplantate die Gesichtssymmetrie weiter verbessern. Die Distraktionsosteogenese, bei der eine Osteotomie durchgeführt wird und eine Distraktions- (Separator-) Vorrichtung an beiden Teilen des Unterkiefers angebracht wird, wird zunehmend verwendet. Die kieferorthopädische Behandlung hilft im Jugendalter bei der Korrektur der Bissanomalie.
Oberkieferhypoplasie
Die Oberkieferhypoplasie ist eine Unterentwicklung der Oberkieferknochen, die eine Mittelgesichtsretrusion verursacht und die Illusion von Protuberanzen (nach vorne ragend) des Unterkiefers erzeugt.