




Das familiäre Mittelmeerfieber ist eine autosomal-rezessive Erbkrankheit, die durch wiederholte Fieberanfälle und Peritonitis, manchmal mit Pleuritis, Hautläsionen, Arthritis und selten Perikarditis gekennzeichnet ist. Eine renale Amyloidose kann sich entwickeln, die manchmal zu einem Nierenversagen führt. Menschen mit genetischen Ursprung im Mittelmeerraum sind häufiger als andere ethnische Gruppen betroffen. Die Diagnose wird vor allem klinisch gestellt, obwohl inzwischen eine genetische Untersuchung möglich ist. Die Behandlung besteht aus einer prophylaktischen Gabe von Colchicin, die bei den meisten Patienten die akuten Anfälle sowie eine renale Amyloidose verhindern kann. Die Prognose ist mit Behandlung ausgezeichnet.
Das familiäre Mittelmeerfieber (FMF) tritt vornehmlich bei Menschen auf, die ihren genetischen Ursprung im Mittelmeerraum haben, so etwa die sephardischen Juden, die nordafrikanischen Araber, Armenier, Türken, Griechen und Italiener. Es gibt jedoch auch genügend Fälle bei vielen anderen ethnischen Gruppen (z. B. Ashkenazi-Juden, Kubaner, Belgier), sodass Vorsicht geboten ist und die Diagnose nicht allein aufgrund der Herkunft ausgeschlossen werden sollte. Ungefähr 50% der Patienten haben eine positive Familienanamnese, meistens sind die Geschwister betroffen.
Ätiologie
Familiäres Mittelmeerfieber wird verursacht durch
Mutationen im MEFV Gen am kurzen Arm von Chromosom 16
Die Mutation wird auf autosomal rezessive Weise vererbt. FMF-Mutationen sind Funktionsgewinn, das heißt, sie verleihen einem Protein neue oder verstärkte Aktivität mit einer Gendosiswirkung (d. h. mehr Kopien des anormalen Gens vermitteln eine größere Wirkung). Das MEFV-Gen kodiert normalerweise für ein Protein (Pyrin oder Marenostrin), das in den zirkulierenden Neutrophilen exprimiert wird.
Pyrin ist Teil des angeborenen Immunsystems und schützt vor bakteriellen Toxinen, die Actin depolymerisieren und Inflammasome aktivieren. Bakterien, die solche Toxine produzieren, schließen Clostridioides difficile, Burkholderia cenocepacia, und Vibrio cholerae (1) ein. Es wird vermutet, dass Pyrin die Entzündungsreaktion stumpf macht, möglicherweise durch Hemmung der Neutrophilenaktivierung und Chemotaxis. Genmutationen führen zu veränderten Pyrinmolekülen, die die Aktivierung von Entzündungskrankheiten nicht hemmen und somit kleine, unbekannte Entzündungsauslöser nicht unterdrücken können, die normalerweise von intaktem Pyrin überprüft werden. Die klinische Folge ist ein spontaner Fieberanfall mit einer hauptsächlich neutrophilen Entzündung im Abdominalraum sowie an anderen Stellen. Es gibt deutliche Hinweise darauf, dass Yersinia pestis, der Erreger der Beulenpest, zur positiven Selektion von FMF-assoziierten MEFV-Mutationen geführt hat. Diese Mutationen verschaffen bestimmten Menschen, die Yersinia pestis beherbergen, einen Überlebensvorteil (2).
Literatur zur Ätiologie
1. Park YH, Wood G, Kastner DL, Chae JJ: Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17(8):914–921, 2016. doi: 10.1038/ni.3457
2. Park YH, Remmers EF, Lee W, et al: Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol 21(8):857–867, 2020. doi: 10.1038/s41590-020-0705-6
Symptome und Beschwerden
Der Beginn des familiären Mittelmeerfiebers liegt in der Regel zwischen 5 und 15 Jahren, kann aber viel später oder früher auftreten, auch im Kindesalter. Die Anfälle treten unregelmäßig auf. Sie dauern 24–72 Stunden, in manchen Fällen sogar länger. Die Häufigkeit der Anfälle schwankt zwischen zwei Anfällen pro Woche bis zu einem Anfall pro Jahr (in den meisten Fällen tritt alle 2–6 Wochen ein Anfall auf). Schweregrad und Häufigkeit nehmen mit zunehmendem Alter, während der Schwangerschaft oder bei Patienten mit einer Amyloidose oft ab. Spontanremissionen können über mehrere Jahre anhalten.
Das Hauptsymptom ist Fieber bis zu 40 °C, meist begleitet von einer Peritonitis. Bei 95% der Patienten kommt es zu Bauchschmerzen (die meist in einem Quadranten beginnen und sich dann über den ganzen Bauchraum ausbreiten), die in ihrem Schweregrad von Anfall zu Anfall variieren können. Auf dem Höhepunkt eines Anfalls kann es zu Symptomen wie abgeschwächten Darmgeräuschen, geblähtem Abdomen, Abwehrspannung und reaktiver Schmerzhaftigkeit kommen, die bei der körperlichen Untersuchung nicht von einer Darmperforation zu unterscheiden sind. Deshalb werden manche Patienten einer Notlaparotomie unterzogen, bevor die richtige Diagnose gestellt wird. Bei Zwerchfellbeteiligung kann es zu Engegefühl im Thorax und zu Schmerzen in einer oder beiden Schultern kommen.
Weitere Symptome von FMF sind akuter pleuritischer Schmerz (bei 30%), Arthritis (bei 25%) im Bereich der Knie, Knöchel und Hüfte, erysipelähnliche Hautrötungen im Bereich des Unterschenkels, Schwellungen des Hodensacks und Schmerzen durch die Entzündung der Tunica vaginalis der Hoden. Eine rezidivierende Perikarditis mit Brustschmerzen ist selten. Die pleuralen, synovialen und dermalen Manifestationen des FMF variieren in ihrer Häufigkeit bei den verschiedenen Bevölkerungsgruppen und sind in den USA seltener anzutreffen als in anderen Ländern.
Trotz der Schwere der Symptome im akuten Anfall erholen sich die meisten Patienten rasch und bleiben bis zum nächsten Anfall krankheitsfrei.
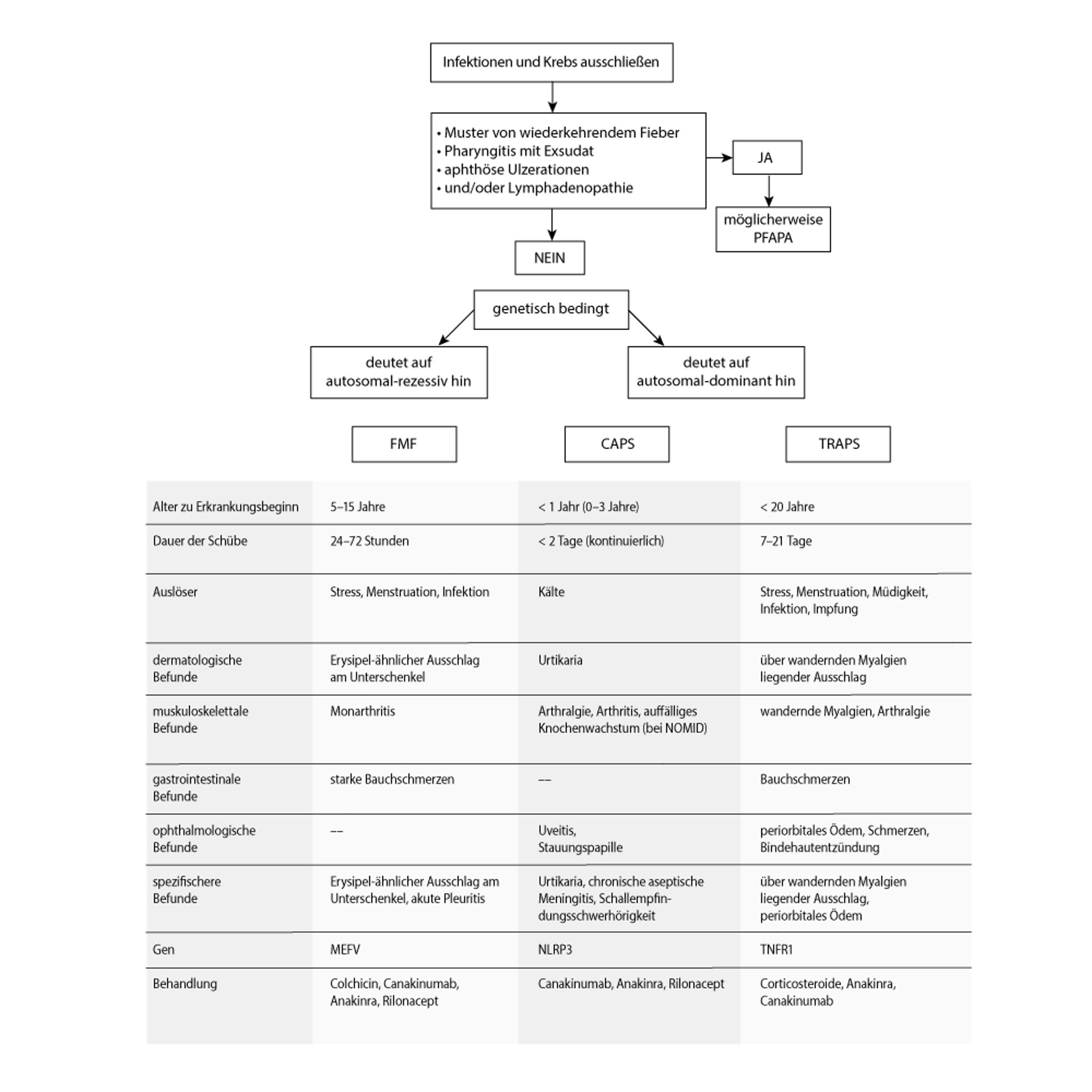
Autoinflammatorische periodische Fiebererkrankungen
CAPS = Kryopyrin-assoziierte periodische Syndrome; FMF = familiäres Mittelmeerfieber; NOMID = neonatal einsetzende multisystemale entzündliche Erkrankung; PFAPA = periodisches Fieber mit Aphthenstomatitis, Pharyngitis und Adenitis; TRAPS = Tumornekrosefaktor-Rezeptor-assoziiertes periodisches Syndrom. Adapted from Sag E, Bilginer Y, Ozen S: Autoinflammatory diseases with periodic fevers. Curr Rheumatol Rep 19(7):41, 2017. doi: 10.1007/s11926-017-0670-8 |

Komplikationen des familiären Mittelmeerfiebers
Die bedeutendste Langzeitkomplikation von FMF ist
Chronische Niereninsuffizienz, die durch die Ablagerungen des Amyloids in den Nieren verursacht wird.
Amyloid kann sich auch im Gastrointestinaltrakt, in der Leber, im Herz, in den Hoden und in der Schilddrüse ablagern.
Familiäres Mittelmeerfieber verursacht Unfruchtbarkeit oder Fehlgeburt bei etwa einem Drittel der Frauen, weil sich Geschwulste bilden, die die Konzeption behindern. Bei Frauen mit familiärem Mittelmeerfieber enden ca. 20–30% der Schwangerschaften in einer Fehlgeburt.
FMF erhöht das Risiko für andere entzündliche Erkrankungen, wie z. B. ankylosierende Spondylitis, Immunglobulin-A-assoziierte (IgA) Vaskulitis (früher Purpura Henoch-Schönlein), juvenile idiopathische Arthritis, Polyarteriitis nodosa, multiple Sklerose und Behçet-Krankheit (1).
Hinweise auf Symptome und Zeichen
1. Balcı-Peynircioğlu B, Kaya-Akça Ü, Arıcı ZS, et al: Comorbidities in familial Mediterranean fever: Analysis of 2000 genetically confirmed patients. Rheumatology (Oxford) 59(6):1372–1380, 2020. doi: 10.1093/rheumatology/kez410. PMID: 31598713
Diagnose
Klinische Abklärung
Gentests
Die Diagnose von familiärem Mittelmeerfieber wird hauptsächlich klinisch basierend auf Tel HaShomer-Kriterien gestellt (siehe Tabelle Tel HaShomer Criteria for the Diagnosis of Familial Mediterranean Fever; 1), aber Gentests sind verfügbar und sind besonders nützlich bei der Bewertung atypischer Fälle. Aktuelle Gentests sind jedoch nicht unfehlbar; einige Patienten mit phänotypisch unverwechselbarem FMF haben nur ein einziges mutiertes Gen oder gelegentlich keine offensichtlichen Mutationen im MEFV-Gen. Etwa 10 bis 20% der Patienten, die die diagnostischen Kriterien für FMF erfüllen, weisen keine MEFV-Mutationen auf, was darauf hindeutet, dass epigenetische und umweltbedingte Faktoren zur Pathogenese der Krankheit beitragen (2).
Unspezifische Befunde sind erhöhte Leukozyten mit einem Überwiegen der Segmentkernigen, eine erhöhte Erythrozytensedimentationsrate, CRP und erhöhtes Fibrinogen. Eine Urinausscheidung von > 0,5 g Eiweiß/24 h weist auf eine renale Amyloidose hin.
Die Differenzialdiagnose sollte eine akute intermittierende Porphyrie, ein hereditäres Angioödem mit abdominalen Anfällen, eine Pankreatitis und andere erbliche Arten eines erneuten Fieberanfalls einschließen.
Literatur zur Diagnose
1. Livneh A, Langevitz P, Zemer D, et al: Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40(10):1879–1885, 1997. doi: 10.1002/art.1780401023
2. Booty MG, Chae JJ, Masters SL, et al: Familial Mediterranean fever with a single MEFV mutation: Where is the second hit? Arthritis Rheum 60(6):1851–1861, 2009. doi: 10.1002/art.24569
Behandlung
Colchicin
Manchmal Canakinumab, Anakinra oder Rilonacept
Durch die prophylaktische Gabe von Colchicin 0,6 mg p.o. 2-mal täglich (manche Patienten benötigen eine -mal tägliche Dosierung, andere eine Einzeldosis) kommt es bei 85% der Patienten zur vollständigen Remission oder zu einer deutlichen Besserung. Wenn Anfälle oder subklinische Entzündungen fortbestehen, sollte die Colchicin-Dosis erhöht werden. Bei Patienten mit seltenen Anfällenmit seltenen Attacken, die einen allmählichen Beginn haben, kann Colchicin nach Auftreten der ersten Symptome gegeben werden, anfangs 0,6 mg p.o. stündlich über 4 h, dann alle 2 h über 4 h, dann alle 12 h über 48 h. Der Einsatz von Colchicin auf dem Höhepunkt eines Anfalls hat selbst bei IV Applikation kaum eine positive Wirkung. Kinder benötigen oft eine Erwachsenendosis, um eine effektive Prophylaxe zu erreichen. Die breite, prophylaktische Anwendung von Colchicin ist mit einem dramatischen Rückgang der Inzidenz von Amyloidose und konsekutivem Nierenversagen assoziiert.
Colchicin führt bei den betroffenen Frauen nicht zu einem erhöhten Infertilitäts- und Fehlgeburtsrisiko und erhöht auch nicht die Anzahl teratogener Schädigungen, wenn es in der Schwangerschaft eingenommen wird. Eine schlechte Compliance bei der Medikamenteneinnahme führt oft zu einer fehlenden Reaktion auf Colchicin, es kann aber auch eine schlechte Korrelation vorliegen, wenn eine verminderte Colchicinkonzentration in den Monozyten vorliegt.
Alternative Therapien für Nichtansprechende sind Anakinra 100 mg subkutan einmal täglich, Rilonacept 2,2 mg/kg subkutan wöchentlich oder Canakinumab 150 mg subkutan alle 4 Wochen (1, 2).
Zur Schmerzlinderung müssen manchmal Opioide eingesetzt werden. Es sollte jedoch wegen einer möglichen Abhängigkeit vorsichtig damit umgegangen werden.
Literatur zur Behandlung
1. Ozen S, Demirkaya E, Erer B, et al: EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis 75(4):644–651, 2016. doi: 10.1136/annrheumdis-2015-208690
2. De Benedetti F, Gattorno M, Anton J, et al: Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med 378(20):1908–1919, 2018. doi: 10.1056/NEJMoa1706314
Wichtige Punkte
Familiäres Mittelmeerfieber wird durch eine autosomal-rezessive Mutation in einem Protein hervorgerufen, das dafür da ist, die entzündliche Reaktion in den Neutrophilen zu modulieren.
Menschen mit genetischen Ursprung im Mittelmeerraum sind häufiger (aber nicht ausschließlich) betroffen.
Die Patienten haben kurze Anfälle von Fieber, Bauchschmerzen und manchmal auch andere Symptome wie Pleuritis, Arthritis und Hautausschlag.
Die renale Amyloidose, die manchmal zu Nierenversagen führt, ist die häufigste Komplikation. Eine prophylaktische Colchicingabe bietet jedoch Schutz gegen die Amyloidose.
Die Diagnose wird klinisch gestellt, bei atypischen Fällen sollten jedoch Gentests zum Einsatz kommen.
Tägliches Colchicin führt bei den meisten Patienten zu einem erheblichen Schutz gegen Anfälle, aber einige erfordern auch einen Immunmodulator wie Anakinra, Rilonacept oder Canakinumab.