




Sarkoidose ist eine entzündliche Erkrankung, die durch nichtverkäsende Granulome in einem oder mehreren Organen und Geweben entsteht; die Ätiologie ist unbekannt. Am häufigsten sind Lunge und Lymphsystem betroffen, die Sarkoidose kann jedoch alle Organe befallen. Die pulmonale Symptomatik variiert von asymptomatisch bis hin zu Husten, Belastungsdyspnoe und selten Lungeninsuffizienz oder Versagen anderer Organsysteme. Die Diagnose wird normalerweise primär wegen der Lungenbeteiligung vermutet und durch einen Röntgenthorax, eine Biopsie und den Ausschluss anderer granulomatöser Entzündungen bestätigt. Die Behandlung ist in der Regel bei symptomatischen Patienten indiziert. First-line-Medikamente sind Kortikosteroide. Die Prognose ist bei begrenzter Ausdehnung („limited disease“) ausgezeichnet, im weiter fortgeschrittenen Stadium („advanced disease“) jedoch schlecht.
Die Sarkoidose tritt meist im Alter zwischen 20 und 40 Jahren, gelegentlich jedoch auch bei Kindern und älteren Erwachsenen auf. Weltweit ist die Prävalenz bei schwarzen Amerikanern und ethnischen Nordeuropäern, insb. Skandinaviern, am höchsten. Das klinische Erscheinungsbild variiert je nach ethnischer Zugehörigkeit und ethnischem Hintergrund stark, wobei schwarzamerikanische Patienten häufiger extrathorakale Manifestationen aufweisen. Sarkoidose kommt bei Frauen etwas häufiger vor.
Löfgren-Syndrom
Das Löfgren-Syndrom manifestiert sich als eine Triade akuter wandernder Polyarthritis, Erythema nodosum und hilärer Lymphadenopathie. Fieber, Unwohlsein, Uveitis sowie Parotitis können ebenfalls vorhanden sein. Das Löfgren-Syndrom tritt häufiger bei Menschen europäischer Abstammung auf. Das Löfgren-Syndrom ist selbstlimitierend. Die Patienten können in der Regel allein mit nichtsteroidalen Antirheumatika (NSAR) behandelt werden. Die Rezidivrate ist gering.
Heerfordt-Syndrom
Das Heerfordt-Syndrom (Uveoparotid-Fieber) manifestiert sich als Schwellung der Ohrspeicheldrüse (aufgrund Sarkoid-Infiltration), Uveitis, chronisches Fieber und seltener Lähmung des N. facialis. Das Heerfordt-Syndrom kann oft selbstlimitierend sein. Die Therapie ist die gleiche wie bei für Sarkoidose.
Blau-Syndrom
Beim Blau-Syndrom handelt es sich um eine sarkoidoseähnliche Erkrankung, die autosomal dominant vererbt wird und sich bei Kindern manifestiert. Es ist nicht bekannt, ob das Blau-Syndrom durch den gleichen Mechanismus entsteht wie die bei Erwachsenen diagnostizierte Sarkoidose. Beim Blau-Syndrom leiden Kinder vor dem 4. Lebensjahr unter Arthritis, Hautausschlag und Uveitis. Das Blau-Syndrom ist oft selbstlimitierend. Die Symptome werden in der Regel mit nichtsteroidalen Antirheumatika (NSAR) gelindert.
Ätiologie der Sarkoidose
Es wird angenommen, dass die Sarkoidose auf eine übertriebene Entzündungsreaktion auf ein Allergen aus der Umwelt bei einer genetisch anfälligen Person zurückzuführen ist. Vorgeschlagene Auslöser sind
Propionibacterium acnes und Mykobakterien (möglicherweise die Mycobacterium tuberculosis Katalase-Peroxidase [mKatG] Protein)
Schimmel oder Mehltau und bestimmte, nicht-identifizierte Substanzen kommen in Arbeitsplätzen mit muffigen Gerüchen und vor.
Pestizide, insbesondere solche, die Aluminiumverbindungen enthalten
Tabakkonsum ist invers korreliert mit Sarkoidose.
Nachweise für genetische Anfälligkeit beinhaltet folgendse:
Höhere Rate der Krankheitskonkordanz bei eineiigen als zweieiigen Zwillingen
Erhöhte Prävalenz der Sarkoidose (etwa 3,6 bis 9,6%) unter Verwandten 1.-oder 2. Grades von Patienten mit Sarkoidose
Verfünffachung des relativen Risikos für Sarkoidose bei Geschwistern von Patienten, die Sarkoidose haben
Identifizierung mehrerer möglicher humaner Leukozyten-Antigene (HLA) und Nicht-HLA-Gene, die mit Sarkoidose-Risiko, dem Verlauf und den Phänotypen in Verbindung stehen
So ist beispielsweise der HLA-DRB1*03/DQB1*02-Haplotyp mit dem Löfgren-Syndrom assoziiert und sagt eine ausgezeichnete Prognose voraus, im Gegensatz zu HLA-DRB1*15/HLA DQB1*0602, der eine persistierende Erkrankung voraussagt.
Pathophysiologie der Sarkoidose
Die unbekannten Auslöser verursachen eine zellvermittelte Immunantwort, die durch eine Akkumulation von T-Zellen und Makrophagen, die Freisetzung von Zytokinen und Chemokinen und die Organisation der an der Antwort beteiligten Zellen zu Granulomen gekennzeichnet ist. Gehäuftes Auftreten innerhalb von Familien und Bevölkerungsgruppen lässt eine genetische Prädisposition, eine gemeinsame Exposition oder – was allerdings weniger wahrscheinlich ist – eine Übertragung von Mensch zu Mensch vermuten.
Der Entzündungsprozess Führt in die Entwicklung nichtverkäsender Granulome, das pathologische Kennzeichen der Sarkoidose. Granulome sind Ansammlungen von mononukleären Zellen und Makrophagen, die unterschieden werden in Epitheloidzellen und mehrkernigen Riesenzellen und von Lymphozyten, Plasmazellen, Fibroblasten und Kollagen umgeben sind. Granulome treten am häufigsten in der Lunge und den Lymphknoten auf, können aber jedes Organ betreffen und zu erheblichen Funktionsstörungen führen. Granulome in der Lunge sind entlang den Lymphgefäßen in peribronchiolärer, subpleuraler und perilobulärer Häufung angeordnet. Die Akkumulation von Granulomen verzerrt die Architektur in den betroffenen Organen. Ob Granulome direkt zur Fibrose führen oder einen parallelen Verlauf nehmen, ist nicht bekannt.
Eine Hyperkalzämie kann aufgrund einer erhöhten Umwandlung von Vitamin D in die aktivierte Form (1,25-Hydroxy-Vitamin D) durch Makrophagen auftreten. Hypercalciurie kann vorhanden sein, auch bei Patienten mit normalen Serumkalziumspiegeln. Nephrolithiasis und Nephrokalzinose können auftreten und manchmal zu chronischen Nierenerkrankungen führen.

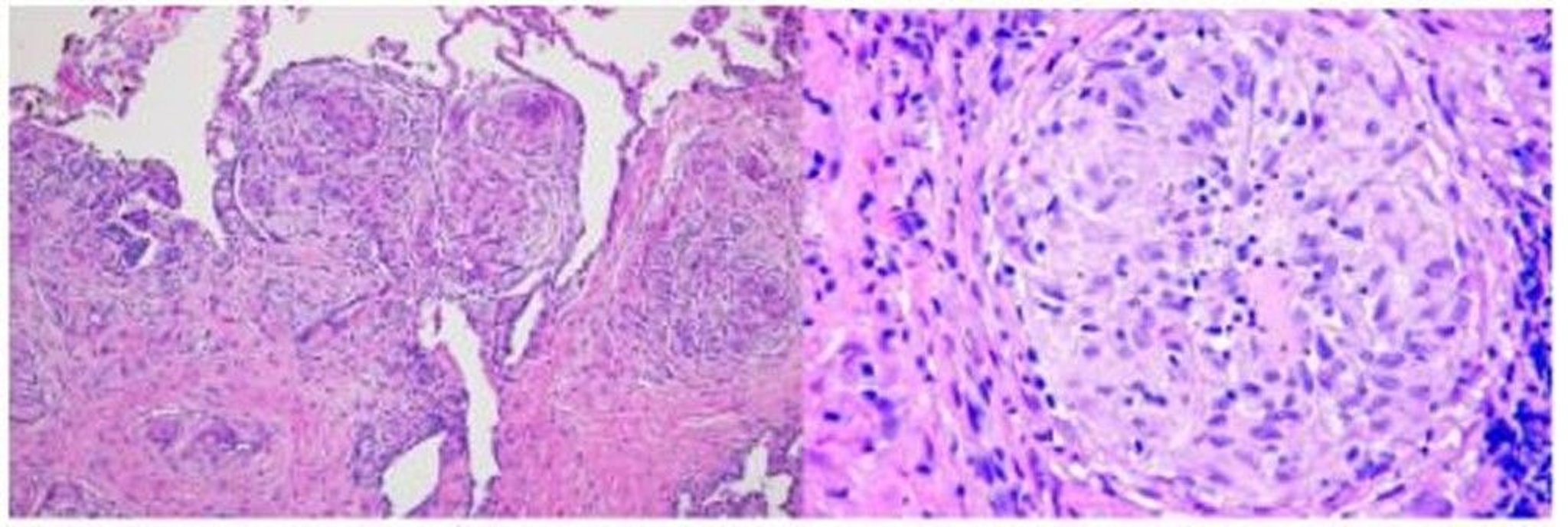
Image courtesy of Birendra P. Sah, MD, FCCP.
Symptome und Anzeichen von Sarkoidose
Die Symptome und Beschwerden sind vom Manifestationsort und Ausmaß der Erkrankung abhängig, verändern sich im Laufe der Zeit, und es zeigen sich Spontanremissionen bis hin zu chronisch indolenten Erkrankungen. Dementsprechend ist eine häufige Neubewertung im Hinblick auf neue Symptome und die Beteiligung verschiedener Organe erforderlich. Die meisten Fälle verlaufen vermutlich asymptomatisch und bleiben daher unentdeckt. Lungenbeteiligung tritt bei > 90% der erwachsenen Patienten auf.
Die Symptomatik kann aus Dyspnoe, Husten, thorakalem Engegefühl und Rasselgeräuschen bestehen. Müdigkeit, Krankheitsgefühl, Schwäche, Kachexie, Gewichtsverlust und leichtes Fieber treten ebenfalls häufig auf. Eine Sarkoidose kann sich als Fieber ungeklärter Ursache manifestieren. Systemische Beteiligung führt zu verschiedenen Symptomen (siehe Tabelle Systemische Beteiligung an Sarkoidose), die sich je nach Rasse, Geschlecht und Alter unterscheiden. Schwarze zeigen häufiger als Weiße eine Beteiligung von Augen, Leber, Knochenmark, peripheren Lymphknoten und der Haut; Erythema nodosum ist die Ausnahme. Frauen leiden mit höherer Wahrscheinlichkeit an einem Erythema nodosum sowie einer Beteiligung von Auge oder Nervensystem. Bei Männern und älteren Patienten ist die Wahrscheinlichkeit einer Hyperkalzämie größer.
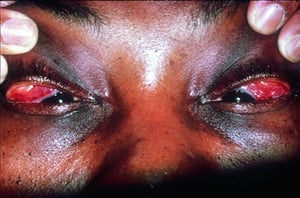
© Springer Science+Business Media
© Springer Science+Business Media
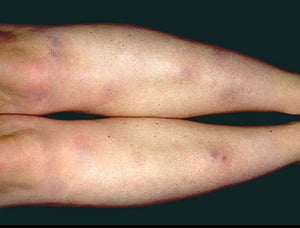
Image provided by Thomas Habif, MD.
© Springer Science+Business Media
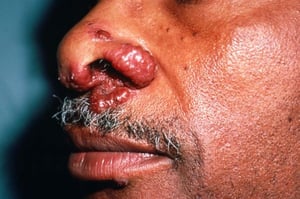
Image courtesy of Karen McKoy, MD.
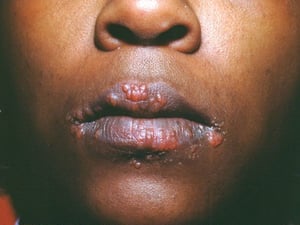
Image courtesy of Karen McKoy, MD.
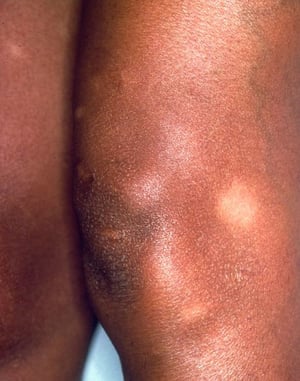
Image courtesy of Karen McKoy, MD.


© Springer Science+Business Media

© Springer Science+Business Media

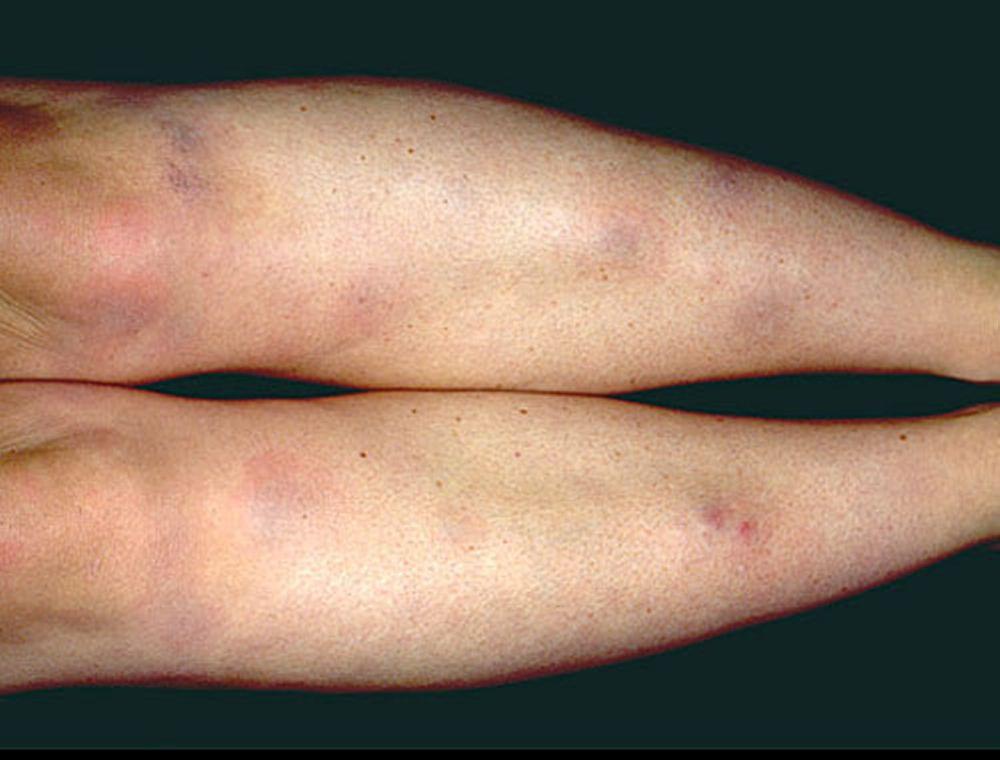
Image provided by Thomas Habif, MD.

© Springer Science+Business Media

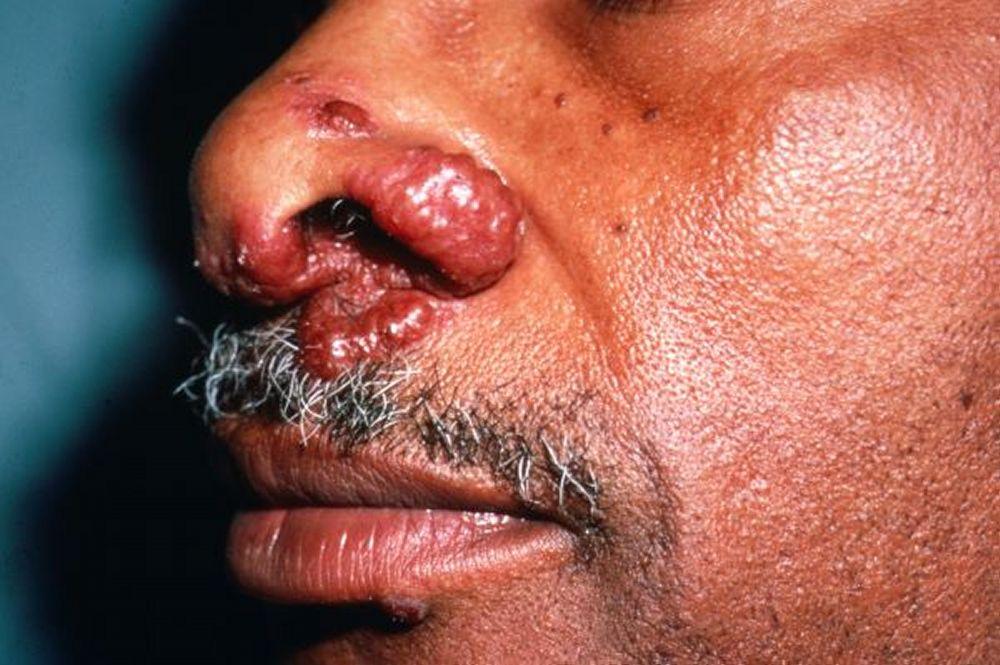
Image courtesy of Karen McKoy, MD.

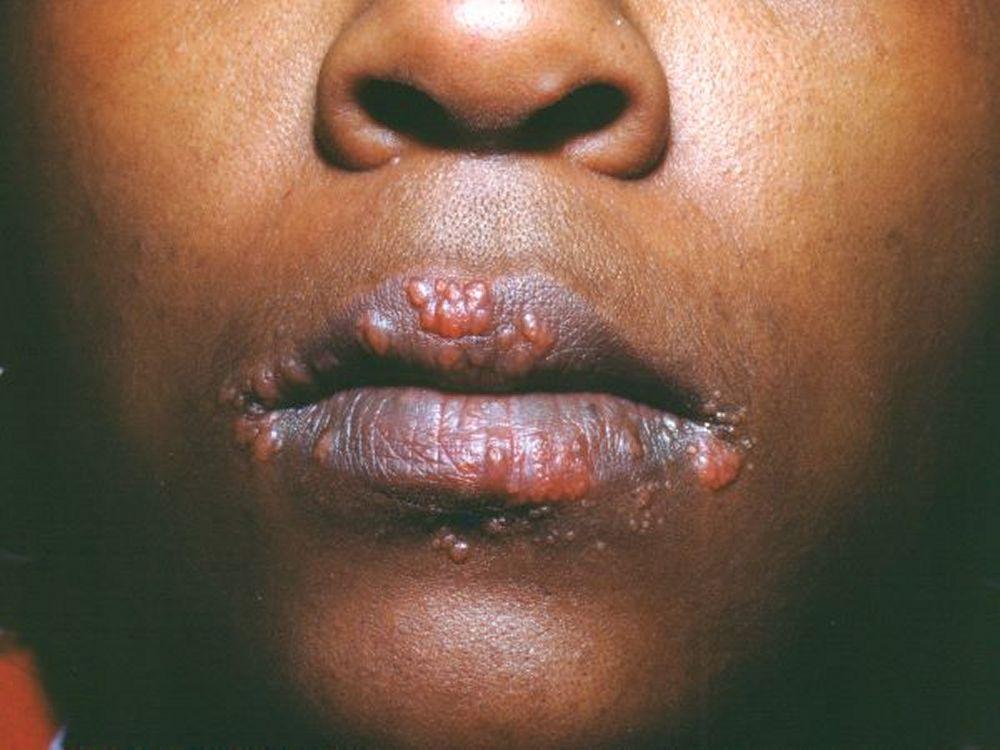
Image courtesy of Karen McKoy, MD.

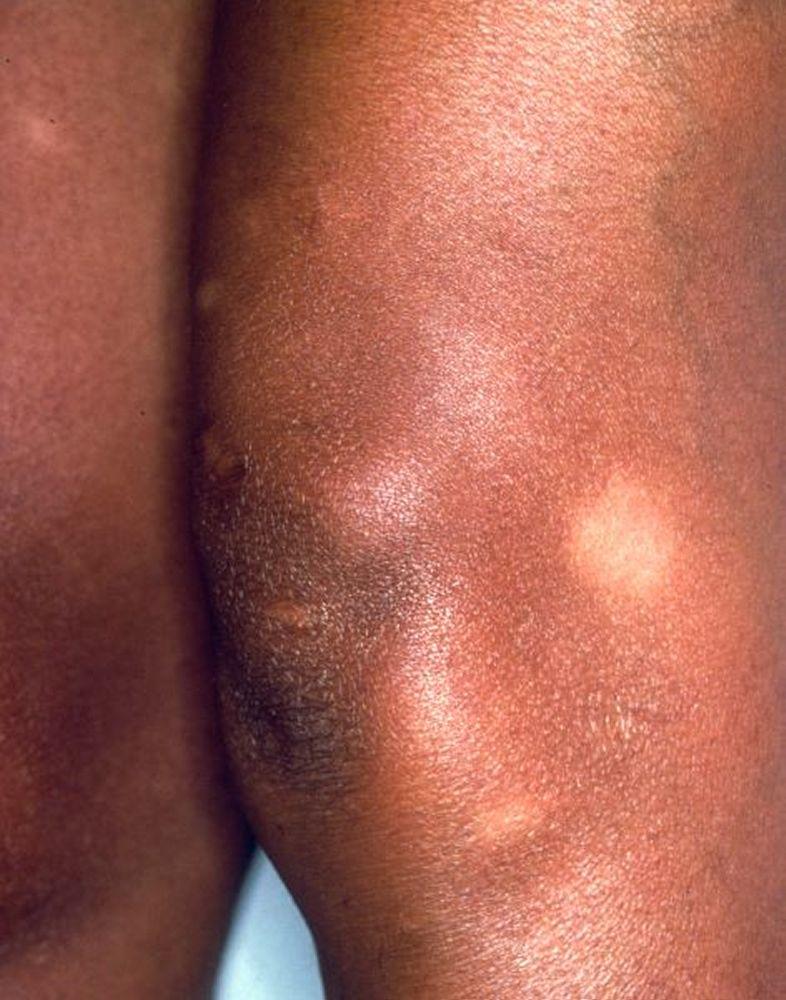
Image courtesy of Karen McKoy, MD.
Kinder mit Sarkoidose können das Blau-Syndrom (Arthritis, Hautausschlag, Uveitis) aufweisen oder Manifestationen ähnlich denen von Erwachsenen haben. In dieser Altersgruppe kann die Sarkoidose mit einer juvenilen idiopathischen Arthritis (juvenile rheumatoide Arthritis) verwechselt werden.
Diagnose von Sarkoidose
Bildgebende Verfahren
Biopsie
Ausschluss anderer granulomatöser Erkrankungen
Eine Sarkoidose wird am häufigsten vermutet, wenn eine hiläre Adenopathie mit oder ohne Lungeninfiltrate zufällig im Röntgenthorax entdeckt wird. Bilaterale Hilaradenopathie ist die häufigste Anomalie.
Bei Verdacht auf Sarkoidose sollte ein Röntgenthorax die erste Maßnahme sein, wenn nicht bereits passiert. Das Röntgenbild neigt dazu, die Wahrscheinlichkeit einer Spontanremission (siehe Tabelle Röntgenthorax zur Stadieneinteilung bei Sarkoidose) bei Patienten mit ausschließlich thorakaler Lymphknotenbeteiligung grob vorherzusagen. Doch die Feststellung von Sarkoidose durch Röntgenthorax kann irreführend sein; z. B. kann eine extrapulmonale Sarkoidose, wie Herzsarkoidose oder neurologische Sarkoidose, eine schlechte Prognose ohne Beweis für Lungenbeteiligung bedeuten. Außerdem sagen Ergebnisse von Röntgenaufnahmen der Brust die Lungenfunktion nur schlecht voraus, sodass Röntgenaufnahme des Thorax nicht genau den Schweregrad der Lungensarkoidose zeigen.
Eine normale Röntgenaufnahme des Thorax (Stufe 0), schließt nicht die Diagnose einer Sarkoidose aus, insbesondere wenn eine Herz- oder neurologische Beteiligung vermutet wird. Eine hochauflösende CT ist zur Erfassung hilarer und Lymphadenopathie des Mediastinums sowie parenchymaler Anomalien empfindlicher. Das Lungenparenchym ist vor allem in den Oberlappen betroffen, kann aber in jedem Teil der Lunge vorkommen. Zu den CT-Befunden (siehe auch Abbildung Thorax-CT-Scan bei pulmonaler Sarkoidose) in fortgeschrittenen Stadien (II bis IV) gehören die folgenden:
Verdickung der bronchovaskulären Bündel und Bronchialwände
Beading der interlobulären Septen
Milchglastrübungen
Parenchymknoten, Zysten oder Hohlräume
Traktionsbronchiektasen
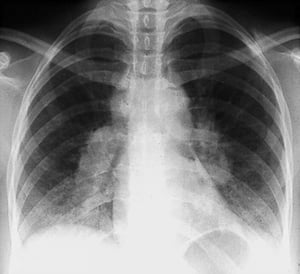
By permission of the publisher. Aus Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.
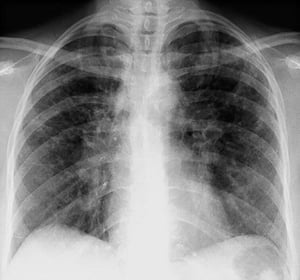
By permission of the publisher. Aus Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.
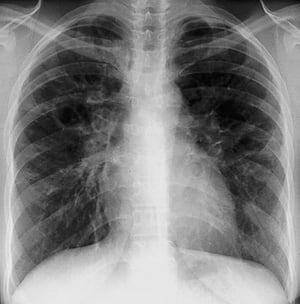
By permission of the publisher. Aus Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.
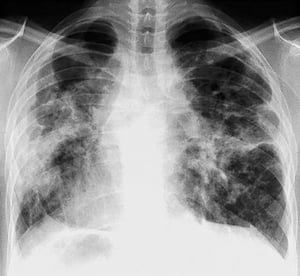
By permission of the publisher. Aus: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.
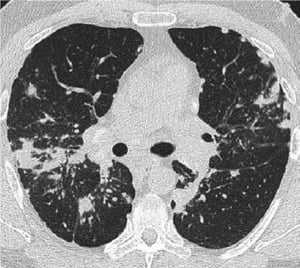
Image courtesy of Birendra P. Sah, MD, FCCP.

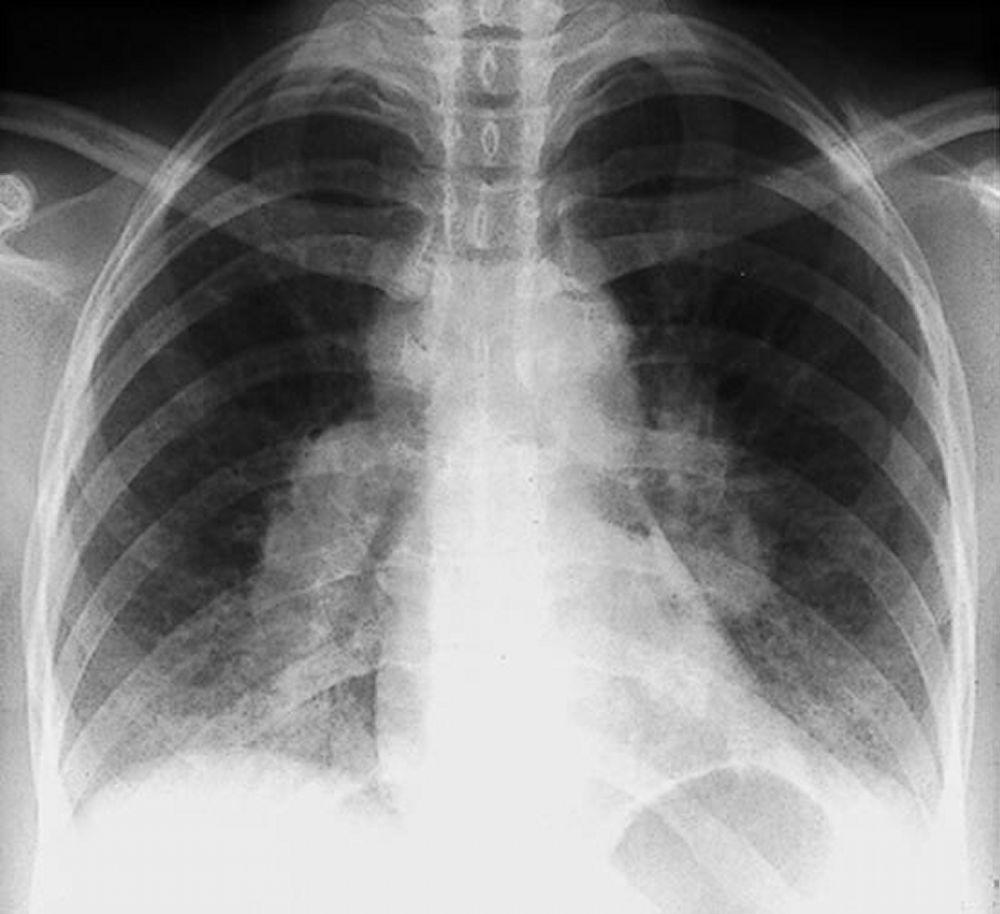
By permission of the publisher. Aus Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.

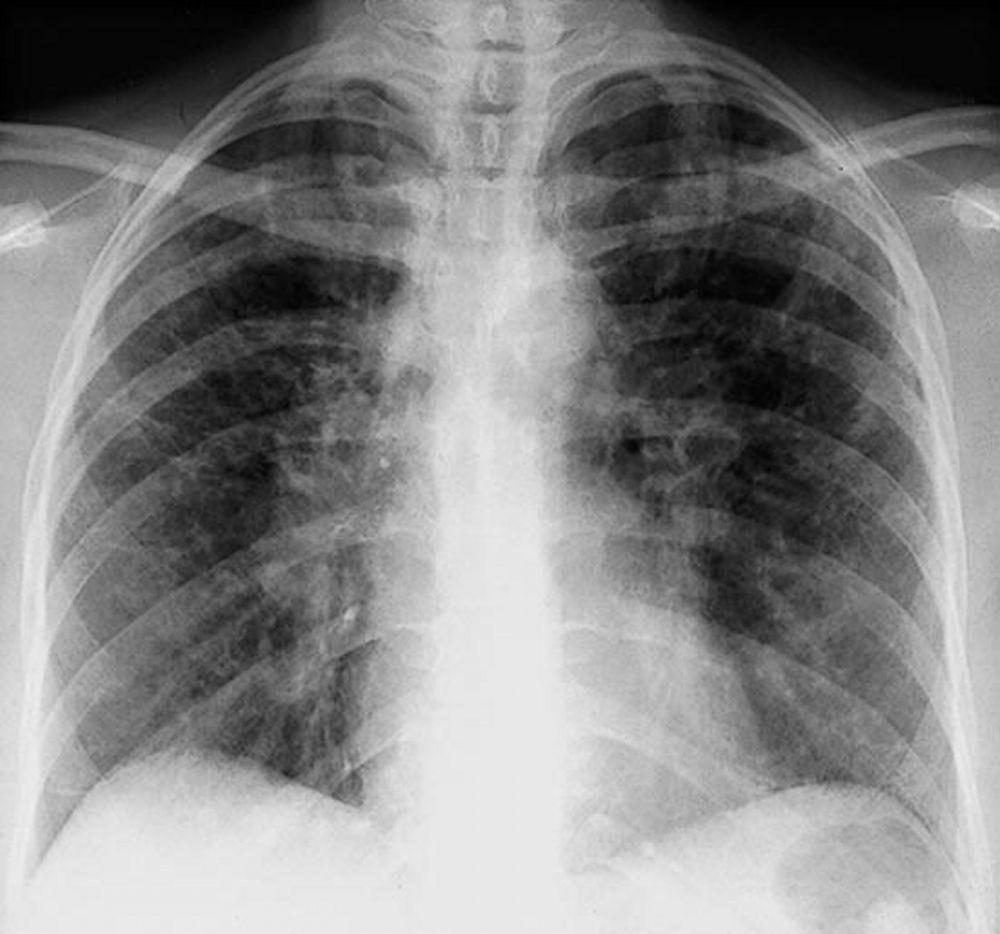
By permission of the publisher. Aus Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.

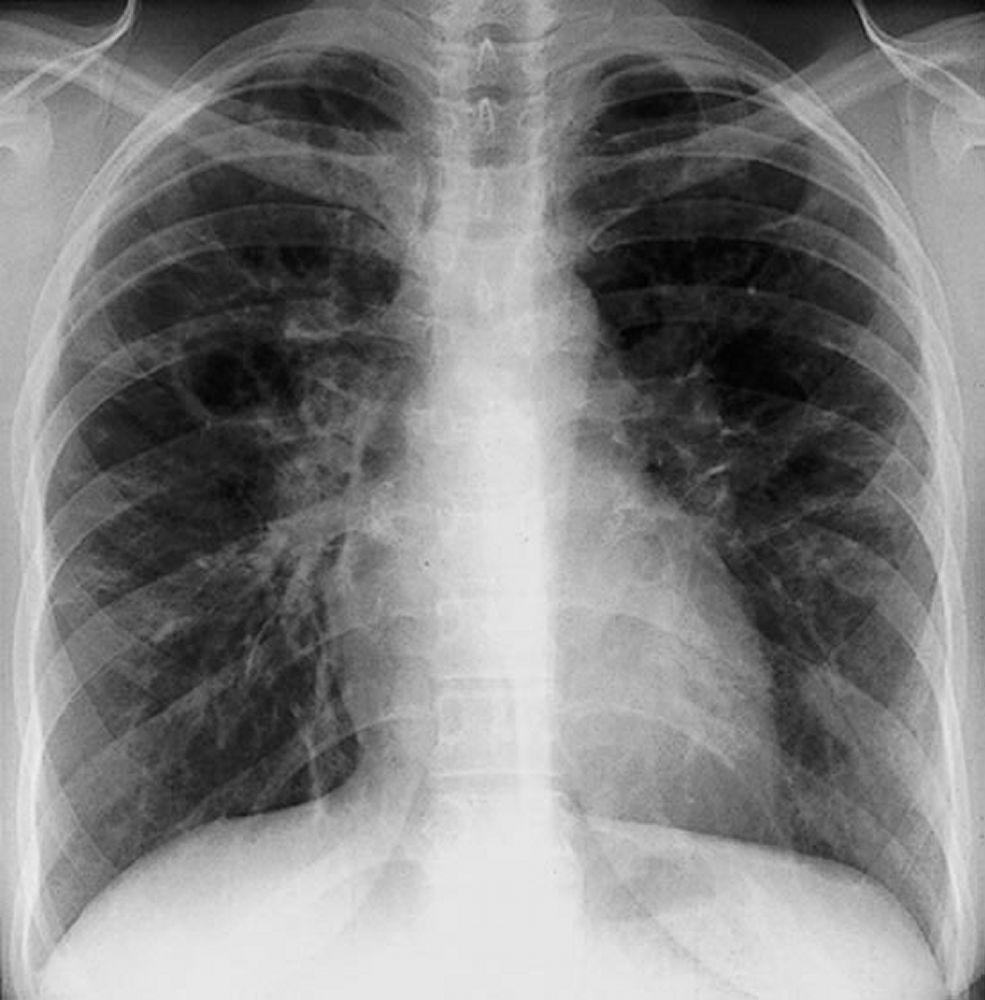
By permission of the publisher. Aus Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.

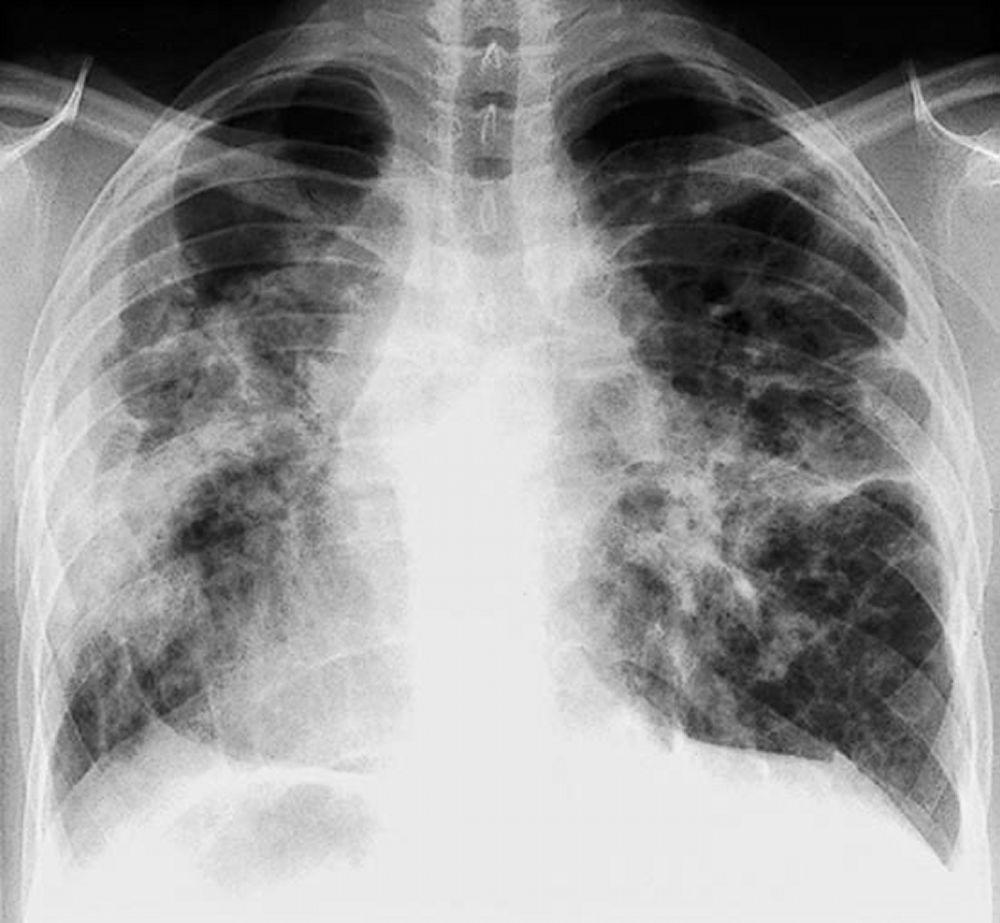
By permission of the publisher. Aus: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Herausgegeben von J Crapo. Philadelphia, Current Medicine, 2005.

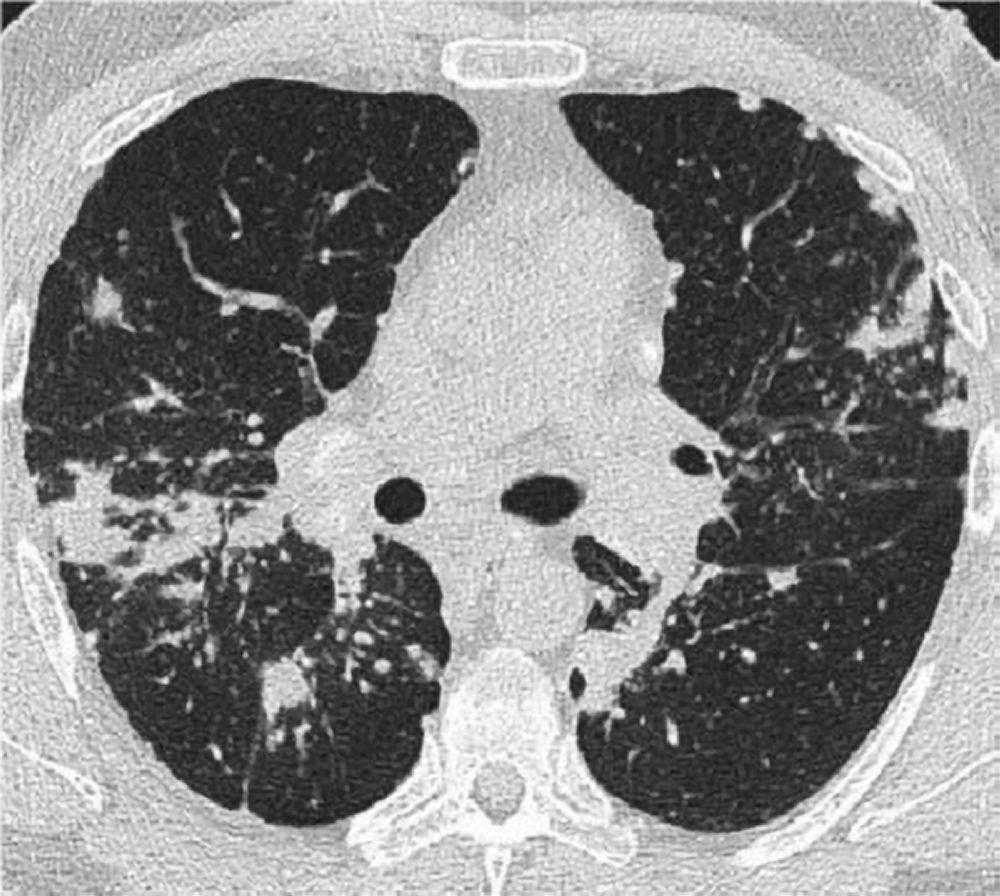
Image courtesy of Birendra P. Sah, MD, FCCP.
Besteht nach Bildgebung weiterhin der Verdacht auf eine Sarkoidose, wird die Diagnose bioptisch durch den Nachweis nichtverkäsender Granulome und durch Ausschluss anderer Ursachen von granulomatösen Erkrankungen (siehe Tabelle Differenzialdiagnose der Sarkoidose) bestätigt. Das Löfgren-Syndrom erfordert keine Bestätigung durch Biopsie.
Die Diagnostik erfordert daher das Folgende:
Auswahl einer Biopsiestelle
Ausschluss anderer Ursachen für granulomatöse Erkrankungen
Beurteilung der Schwere und des Ausmaßes der Erkrankung um festzulegen, ob eine Therapie angezeigt ist
Entnahmestellen für Biopsie
Geeignete Biopsieentnahmestellen können sich während der körperlichen Untersuchung und bei der Erstvorstellung ergeben: periphere Lymphknoten, Hautveränderungen und Konjunktiven sind leicht zugänglich. Eine endobronchial ultraschallgeführte transbronchiale Adelpunktion (EBUS-TBNA) von mediastinalen oder hilaren Lymphknoten hat ein aufgezeigtes diagnostisches Ergebnis von etwa 90% und ist das Diagnoseverfahren der Wahl bei Patienten mit intrathorakalen Beteiligung.
Wenn der Verdacht auf eine pulmonale Sarkoidose besteht und die EBUS-TBNA nicht diagnostisch ist, kann eine bronchoskopische transbronchiale Lungenbiopsie mit bronchoalveolärer Lavage (BAL) durchgeführt werden. Sie kann auch bei Patienten ohne Lungenparenchym-Infiltrate eingesetzt werden, da die diagnostische Ausbeute der transbronchialen Lungenbiopsie bei Sarkoidose im Stadium I etwa 50% beträgt. Wenn die bronchoskopische transbronchiale Biopsie nicht diagnostisch ist, kann sie ein zweites Mal durchgeführt werden.
Wenn EBUS-TBNA und bronchoskopischen transbronchiale Biopsien nicht-diagnostisch sind, oder eine Bronchoskopie nicht toleriert werden kann, kann die Mediastinoskopie durchgeführt werden, um eine Gewebeprobe von mediastinalen oder hilaren Lymphknoten zu entnehmen oder video-assistierte thorakoskopische (VAT) Lungenbiopsie oder offene Lungenbiopsie Biopsie werden kann getan werden, um Lungen erhalten Gewebe. Wenn ein starker Verdacht auf Sarkoidose besteht, aber aufgrund der Untersuchungs- oder Bildgebungsbefunde keine Biopsiestelle erkennbar ist, kann die Positronen-Emissions-Tomographie (PET) helfen, verborgene aktive Stellen wie Herz, Knochen, Muskel und Gehirn zu identifizieren.
Ausschluss anderer Erkrankungen
Der Ausschluss anderer Erkrankungen ist vor allem dann wichtig, wenn Symptomatik und radiologische Veränderungen minimal sind, da viele andere Erkrankungen granulomatöse Entzündungen verursachen können (siehe Tabelle Differenzialdiagnose der Sarkoidose). Biopsiematerial sollte für Pilze und Mykobakterien kultiviert werden. In der Anamnese muss auf eine mögliche Exposition mit beruflichen (z. B. Silica, Beryllium), in der Umwelt vorkommenden (z. B. schimmeliges Heu, Vögel und andere antigene Auslöser einer exogen allergischen Alveolitis) und infektiösen (z. B. Tuberkulose, Kokzidioidomykose, Histoplasmose) Antigenen geachtet werden. Purified protein Derivative (PPD) Hauttests oder Interferon-Gamma-Release-Assay sollten früh im Rahmen der Beurteilung durchgeführt werden.
Beurteilung der Schwere der Erkrankung
Die Bewertung des Schweregrads erfolgt nach Organbeteiligung, z. B. bei alleiniger Beteiligung der Lunge
Lungenfunktionsprüfungen
Im Frühstadium sind die Befunde der Lungenfunktionsprüfung häufig unauffällig, in fortgeschrittenen Stadien sind jedoch Restriktion und eine verminderte CO-Diffusionskapazität (DLCO) zu beobachten. Auch obstruktive Ventilationsstörungen kommen vor und können auf eine Beteiligung der Bronchialschleimhaut hinweisen. Die Durchführung eines 6-Minuten-Gehtests kann die funktionelle Beeinträchtigung umfassender charakterisieren als die Ergebnisse der Lungenfunktionstests allein. Patienten mit ausgedehnter Lungenbeteiligung können in Ruhe eine normale Sauerstoffsättigung aufweisen, bei Anstrengung jedoch eine Entsättigung zeigen.
Empfohlene Routine-Screening-Tests für extrapulmonale Krankheit umfassen
12-Kanal-EKG, Holter-Monitoring und Echokardiographie
Ophthalmologische Spaltlampenuntersuchung
Routine-Bluttests um die Nieren- und Leberfunktion zu bewerten
Serumkalziumspiegel und Kalziumausscheidung im 24-Stunden-Urin
Um eine extrapulmonale Sarkoidose zu erkennen, ist häufig eine Bildgebung erforderlich. Eine kardiale Magnetresonanztomographie (MRT) mit und ohne Gadoliniumkontrast kann bei Patienten mit kardialen Symptomen sinnvoll sein. Bei Patienten mit neurologischen Symptomen kann eine MRT des Gehirns oder der Wirbelsäule mit oder ohne Gadolinium erforderlich sein. Knochenszintigraphien und Elektromyographie können bei Patienten mit rheumatologischen Symptomen angemessen sein. Die PET-Untersuchung scheint der sensitivste Test zum Nachweis von Knochen- und anderer extrapulmonaler Sarkoidose zu sein und wird zusammen mit der MRT bei Patienten mit Herzbeteiligung eingesetzt. Eine Abdomen-CT mit Kontrastmittel wird nicht routinemäßig empfohlen, kann jedoch dem Nachweis einer Leber- oder Milzbeteiligung dienen (z. B. Organvergrößerung, strahlentransparente Läsionen). Eine Ganzkörper-Gallium-Szintigraphie wurde weitgehend durch die Positronenemissionstomographie ersetzt. Falls verfügbar, kann ein Gallium-Scanning nützliche unterstützende Beweise liefern, wenn keine Gewebebestätigung vorliegt. Eine symmetrisch erhöhte Aufnahme in mediastinalen und hilären Lymphknoten (Lambda-Zeichen) sowie in Tränen-, Ohrspeichel- und Speicheldrüsen (Panda-Zeichen) weist stark auf eine Sarkoidose hin. Ein negatives Ergebnis bei Patienten, die Prednison einnehmen, ist nicht aussagekräftig.
Laboruntersuchungen spielen eine ergänzende Rolle bei der Erstellung der Diagnose und der Bestimmung des Ausmaßes der Organbeteiligung. Das vollständige Blutbild mit Differenzialblutbild kann Anämie, Eosinophilie oder Leukopenie zeigen. Serumkalzium sollte gemessen werden, um eine Hyperkalzämie zu erkennen. Harnstoff-, Creatinin- und Leberwerte können bei renaler und hepatischer Sarkoidose ansteigen. Das Gesamt-Eiweiß kann bei Hypergammaglobulinämie erhöht sein. Eine erhöhte Sedimentationsrate der Erythrozyten ist häufig, aber nicht spezifisch. Zum Ausschluss einer Hyperkalziurie wird eine Messung der Kalziumausscheidung im 24 Stunden Sammelurin auch bei normalen Serumkalziumspiegeln empfohlen. Auch erhöhte Angiotension-konvertierenden-Enzym (ACE)-Spiegel können ein Hinweis auf eine Sarkoidose sein. Sie sind jedoch unspezifisch und können bei Patienten mit anderen Erkrankungen ebenfalls erhöht sein (z. B. Hyperthyreose, Diabetes, Morbus Gaucher, Pilzinfektionen, Silikose, Mykobakteriosen, Hypersensitivitätspneumonitis, Lymphome). Erhöhte Spiegel des Angiotensin-konvertierenden Enzyms (ACE) können für die Überwachung der Einhaltung der Kortikosteroidbehandlung nützlich sein. Die ACE-Werte sinken selbst dann ab, wenn die Patienten niedrig dosierte Kortikosteroide einnehmen.
Eine bronchoalveoläre Lavage sollte zusammen mit einer bronchoskopischen Biopsie durchgeführt werden, um den Verdacht auf eine Infektion auszuschließen (z. B. wenn die Ergebnisse weniger invasiver Methoden wie der Bildgebung nicht typisch für eine Sarkoidose sind) und um andere Formen der interstitiellen Lungenerkrankung auszuschließen, wenn die Diagnose einer Sarkoidose zweifelhaft ist. Die BAL-Befunde variieren beträchtlich, aber eine Lymphozytose (Lymphozyten > 15%), ein CD4+/CD8+-Quotient > 3,5 im Differenzialzellbild der Lavageflüssigkeit oder beides weisen im richtigen klinischen Zusammenhang auf die Diagnose hin. Allerdings kann eine Sarkoidose bei Fehlen dieser Befunde nicht ausgeschlossen werden.
Behandlung von Sarkoidose
Nichtsteroidale Antirheumatika (= nonsteroidal anti-inflammatory drugs, NSAID)
Corticosteroide
Immunsuppressiva
Anti-Tumor-Nekrose-Faktor-alpha-Antikörper
Da die Sarkoidose oft spontan ausheilt, benötigen asymptomatische Patienten und Patienten mit leichten Symptomen keine Behandlung. Sie sollten jedoch regelmäßig zum Ausschluss einer Verschlechterung untersucht werden. Diese Patienten können mittels Röntgenuntersuchungen des Thorax, Lungenfunktionsprüfungen (einschließlich Bestimmung der Diffusionskapazität) und Markern der extrathorakalen Beteiligung (z. B. Routineparameter der Nieren- und Leberfunktion, jährliche ophthalmologische Spaltlampenuntersuchung) kontrolliert werden. Die Häufigkeit der Follow-up-Prüfung wird durch den Schweregrad der Erkrankung bestimmt.
Zu den Patienten, die unabhängig vom Thoraxröntgen-Stadium eine Behandlung benötigen, gehören auch Patienten mit folgenden Erkrankungen:
Verschlechterung der Symptome
Einschränkung der Aktivität
Deutlich abnormale oder sich verschlechternde Lungenfunktionen
Besorgniserregende Veränderungen im Röntgenbild (z. B. Kavitation, Fibrose, Konglomeratknoten, Anzeichen pulmonaler Hypertonie)
Herz-, Nervensystem- oder Augenbeteiligung
Nieren- oder Leberversagen
Mittelgradige bis schwere Hyperkalzämie
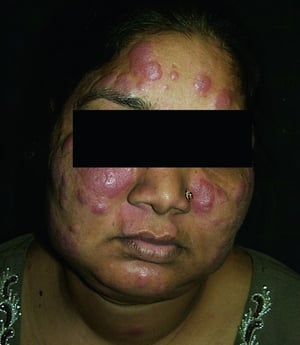
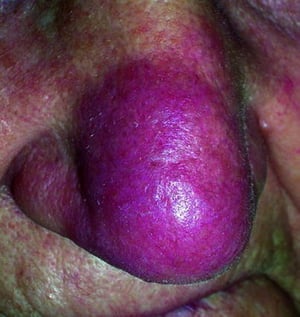
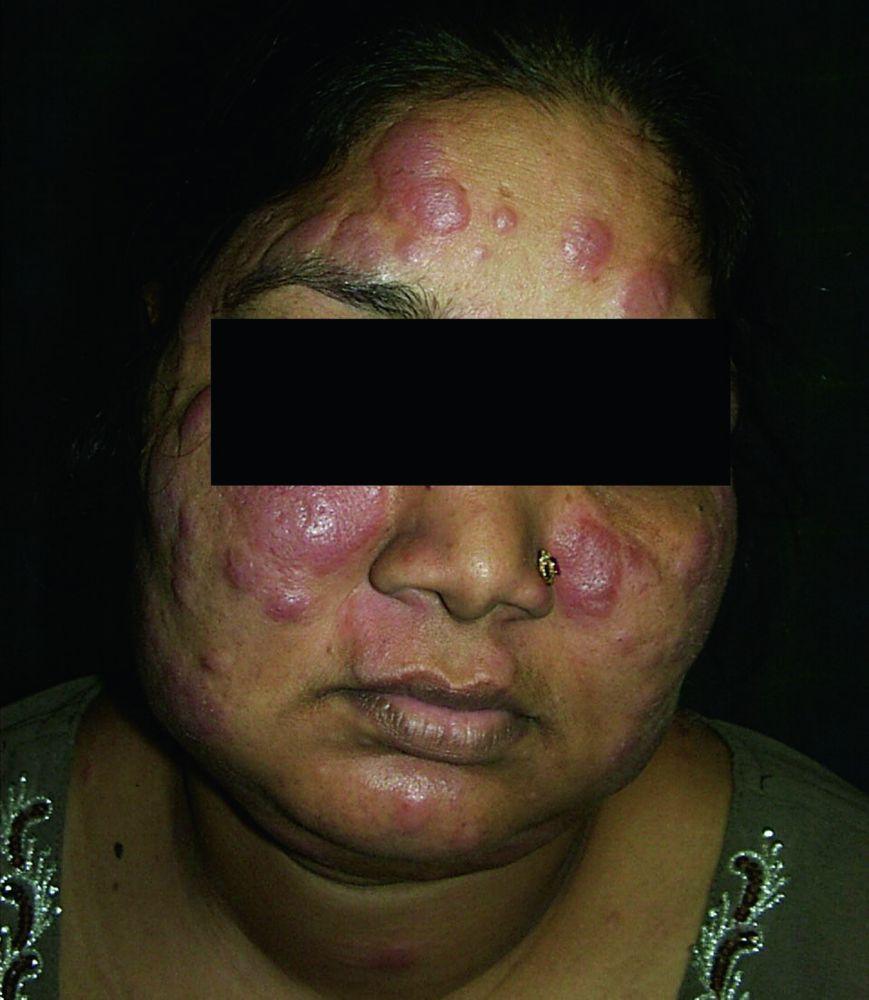
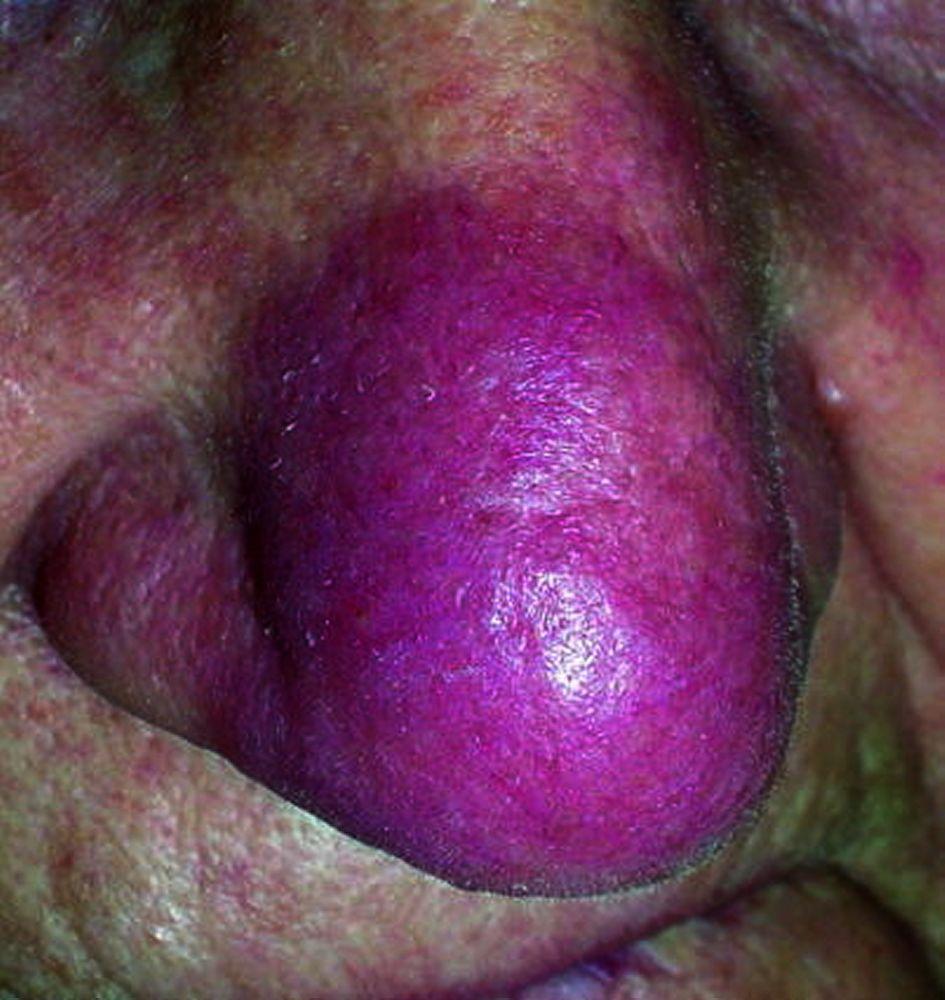
Entstellende Haut- (z. B. Lupus pernio) oder Gelenkerkrankungen
Nichtsteroidale Antirheumatika (NSAR) werden verwendet, um Muskel-Skelett-Schmerzen zu behandeln.
Corticosteroide
Die Symptombehandlung beginnt mit Kortikosteroiden. Das Vorhandensein von Auffälligkeiten im Brustkorb ohne signifikante Symptome oder Anzeichen für eine Abnahme der Organfunktion ist kein Hinweis auf eine Behandlung. Ein Standardprotokoll ist Prednison 20–40 mg oral einmal täglich, je nach Symptomen und Schwere des Befundes. Auch alternierende Schemata können Verwendung finden: z. B. Prednison 40 mg p.o. 1-mal/Tag, jeden zweiten Tag. Obwohl Patienten selten > 40 mg/Tag benötigen, können höhere Dosen erforderlich sein, um Komplikationen bei schwerer neurologischen Erkrankung und Herzerkrankung zu vermeiden. Die Reaktion erfolgt in der Regel innerhalb von 6 bis 12 Wochen, so dass die Symptome, andere Marker für den Schweregrad der Erkrankung und die Ergebnisse von Lungenfunktionstests zwischen 6 und 12 Wochen erneut beurteilt werden können. Chronische, schleichende Fälle können sich langsamer zurückentwickeln. Die Kortikosteroide werden bei Ansprechen bis zur Erhaltungsdosis reduziert (z. B. Prednison 10 bis 15 mg/Tag) und für weitere 6 bis 9 Monate fortgeführt, wenn Besserung eintritt.
Die optimale Therapiedauer ist unbekannt. Frühzeitiges Absetzen kann jedoch zu Rezidiven führen. Das Medikament wird bei fehlendem oder fraglichem Ansprechen langsam ausgeschlichen. Die Kortikosteroide können am Ende bei den meisten Patienten abgesetzt werden. Da jedoch in bis zu 50% der Fälle Rezidive auftreten, sollten Kontrolluntersuchungen normalerweise alle 3–6 Monate wiederholt werden. Die Behandlung mit Kortikosteroiden sollte fortgesetzt werden, wenn Symptome und Beschwerden erneut auftreten. Da die Produktion von Angiotension-Converting-Enzym (ACE) mit niedrigen Dosen von Kortikosteroiden unterdrückt wird, können die seriellen ACE-Spiegel im Serum bei der Beurteilung der Einhaltung von Kortikosteroid-Behandlungen bei Patienten mit erhöhten ACE-Spiegeln nützlich sein.
Inhalative Kortikosteroide können Husten bei Patienten mit endobronchialer Beteiligung lindern. Bei Patienten mit einer obstruktiven Atemwegserkrankung kann ein inhalatives Bronchodilatator hinzugefügt werden.
Topische Kortikosteroide können bei manchen Haut-, Nasennebenhöhlen- und Augenerkrankungen hilfreich sein.
Eine Prophylaxe gegen Pneumocystis jirovecii-Pneumonie ist empfohlen, wenn Patienten länger als einen Monat täglich > 20 mg Prednison oder ein Äquivalent davon einnehmen und bei Patienten, die Immunsuppressiva einnehmen.
Alendronat oder ein anderes Bisphosphonat kann bei Risikopersonen (z. B. älteren Patienten) die Behandlung der Wahl zur Vorbeugung einer durch Kortikosteroide verursachten Osteoporose sein. Bei der Verwendung von Kalzium- oder Vitamin-D-Ergänzungspräparaten besteht die Gefahr einer Hyperkalzämie aufgrund der endogenen Produktion von aktivem Vitamin D (1,25 Dihydroxy-Vitamin D) durch sarkoidale Granulome. Serum- und 24-Stunden-Harn-Kalziummessungen sollten normal sein, bevor mit solchen Ergänzungsmitteln begonnen wird.
Tipps und Risiken
|
Immunsuppressiva
Immunsuppressiva werden verwendet, wenn
Patienten vertragen kein Prednison
die Sarkoidose refraktär ist gegenüber moderaten bis hohen Prednisondosen;
die Prednisondosis nach 3 Monaten nicht unter 10–15 mg täglich gesenkt werden kann.
Vor der Hinzunahme anderer Immunsuppressiva sollten mögliche Gründe für die ausbleibende klinische Verbesserung, wie z. B. Nichteinhaltung, Begleiterkrankungen (z. B. Asthma, Herzinsuffizienz, Anämie), pulmonale Hypertonie und Fibrose im Endstadium, geprüft werden.
Methotrexat ist das am häufigsten verwendete Immunsuppressivum. Bei den Patienten sollte ein 6-monatiger Versuch mit 10–15 mg Methotrexat/Woche unternommen werden. Vor Beginn der Behandlung mit Methotrexat sollten die Patienten auf eine Infektion mit dem Hepatitis-B-Virus und dem Hepatitis-C-Virus getestet werden. Zu Beginn werden MTX und Kortikosteroide überlappend verabreicht, im Laufe von 6 bis 8 Wochen kann das Kortikosteroid reduziert und häufig ganz ausgeschlichen werden. Das maximale Ansprechen auf MTX kann jedoch 6–12 Monate dauern. In diesen Fällen sollte Prednison langsamer ausgeschlichen werden. Kontrollen von Blutbild und Transaminasen sollten zu Beginn alle 2–4 Wochen und nach Erreichen einer gleich bleibenden Erhaltungsdosis alle 6–12 Wochen erfolgen. Für Patienten, die mit Methotrexat behandelt werden, wird Folsäure (1 mg einmal täglich durch den Mund) empfohlen, um das Risiko unerwünschter Wirkungen zu verringern.
Andere Immunsuppressiva sind Azathioprin, Mycophenolat, Cyclophosphamid, Leflunomid und Hydroxychloroquin. Hydroxychloroquin 400 mg p.o. einmal täglich oder 200 mg p.o. zweimal täglich können zur Behandlung von Hyperkalzämie, Gelenkschmerzen, Hautsarkoidose oder vergrößerten unangenehmen oder entstellenden peripheren Lymphknoten wirksam sein. Eine ophthalmologische Untersuchung sollte vor Beginn der Behandlung mit Hydroxychloroquin und alle 6 bis 12 Monate während der Behandlung durchgeführt werden, um die okuläre Toxizität zu überwachen.
Ein Rezidiv ist nach Absetzen eines Immunsuppressivums häufig.
Anti-Tumor-Nekrose-Faktor-alpha-Antikörper
Infliximab wird in der Regel als Medikament der dritten Wahl zur Behandlung der refraktären Sarkoidose und zur Behandlung von Patienten eingesetzt, die sowohl Kortikosteroide als auch die oben genannten Immunsuppressiva nicht vertragen. Vor Beginn der Therapie sollten die Patienten einen Test auf gereinigte Proteinderivate (PPD) oder Interferon-Gamma-Release-Assay durchführen lassen, um auf latente Tuberkulose zu testen. Infliximab wird einmalig intravenös in einer Dosierung von 3–5 mg/kg verabreicht, nach 2 Wochen erneut und dann einmal pro Monat. Es kann 3 bis 6 Monate dauern, bis eine maximale Reaktion erzielt wird. Infliximab wird in der Regel mit niedrig dosiertem Methotrexat oder Azathioprin kombiniert, um die Bildung von Antikörpern gegen das Medikament zu verhindern.
Adalimumab kann bei Patienten mit okulärer Sarkoidose oder Hautsarkoidose und bei Patienten, die erfolgreich mit Infliximab behandelt wurden, aber Antikörper oder Infusionsreaktionen entwickelt haben, in Betracht gezogen werden. Adalimumab 40 bis 80 mg subkutan wird alle 1 bis 2 Wochen verabreicht.
Andere Überlegungen zur Behandlung
Patienten, die einen Herzblock oder ventrikuläre Arrhythmien aufgrund von Herzbeteiligung haben, sollten einen implantierbaren Herzschrittmacher und Defibrillator sowie eine medikamentöse Therapie erhalten.
Mit keinem der zur Verfügung stehenden Medikamente konnte eine Lungenfibrose sicher verhindert werden.
Die Behandlung der Sarkoidose-assoziierten pulmonal-arteriellen Hypertonie ist bei Diurese und zusätzlichem Sauerstoff unterstützend. Die Rolle von pulmonalen Vasodilatatoren bei der Behandlung von SPAH ist nicht eindeutig geklärt; einige kleine Studien lassen auf eine Wirksamkeit schließen, aber es sind größere Studien erforderlich, um diese zu bestätigen (1).
Die Organtransplantation ist eine Möglichkeit für Patienten mit finaler Lungen-, Herz oder Leberbeteiligung; die Erkrankung kann jedoch im transplantierten Organ erneut auftreten.
Bei Patienten mit Sarkoidose und mäßiger oder schwerer Beeinträchtigung der Lungen- oder Herzfunktion besteht ein erhöhtes Risiko für die Entwicklung schwerer Formen der COVID-19-Infektion und der Mortalität. Wie bei Patienten mit anderen entzündlichen Lungenerkrankungen sollten Immunsuppressiva während der COVID-19-Pandemie mit Bedacht eingesetzt werden. Die SARS-CoV-2-Impfung wird bei Patienten mit Sarkoidose dringend empfohlen, da sie die Sterblichkeit und den Schweregrad der Erkrankung aufgrund einer COVID-19-Infektion verringern könnte. Eine laufende klinische Studie soll die Wirksamkeit der SARS-CoV-2-Impfung bei Patienten mit Sarkoidose und bei Patienten, die Immunsuppressiva einnehmen, besser beurteilen.
Literatur zur Therapie
1. Humbert M, Kovacs G, Hoeper MM, et al: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61(1): 1-144, 2023. doi:10.1183/13993003.00879-2022
Prognose bei Sarkoidose
Obwohl Spontanremission häufig ist, sind Krankheitsmanifestationen und Schweregrad sehr variabel, und viele Patienten benötigen Kortikosteroide, um die Symptome zu lindern oder die progressive Organfunktion zu einem Zeitpunkt während des Krankheitsverlaufs zu verlangsamen. Deshalb sind regelmäßige Kontrolluntersuchungen zum Rezidivnachweis obligat. Fast zwei Drittel der Patienten mit Sarkoidose erreichen schließlich eine Remission mit wenigen oder keinen Folgeerscheinungen. Bei ca. 50% der Patienten mit Spontanremission tritt diese innerhalb der ersten 3 Jahre nach Diagnosestellung auf. Weniger als 10% dieser Patienten rezidivieren nach 2 Jahren. Patienten, die innerhalb von 2 bis 3 Jahren keine Remission zeigen, leiden wahrscheinlich an einer chronischen Form.
Sarkoidose soll bei bis zu 30% der Patienten chronisch verlaufen und 10-20% haben permanente Folgeerscheinungen. Die Krankheit ist bei 1 bis 5% der Patienten tödlich, in der Regel aufgrund respiratorischer Insuffizienz wegen einer Lungenfibrose, seltener wegen Lungenblutung, die durch ein Aspergillom verursacht worden ist. Jedoch in Japan ist die häufigste Todesursache eine infiltrative Kardiomyopathie, die zu Arrhythmien und Herzinsuffizienz führt.
Patienten mit extrapulmonaler Sarkoidose und Schwarze haben eine schlechtere Prognose. Eine Remission tritt bei 89% der weißen und 76% der schwarzen Patienten ohne extrathorakale Erkrankung und bei 70% der weißen und 46% der schwarzen Patienten mit extrathorakaler Erkrankung auf.
Gute prognostische Zeichen umfassen
Löfgren-Syndrom (Trias der akuten Polyarthritis, Erythema nodosum und hiläre Lymphadenopathie)
Ungünstige prognostische Zeichen umfassen
Chronische Uveitis
Lupus pernio
Chronische Hyperkalzämie
Neurosarkoidose
Herzbeteiligung
Ausgedehnte pulmonale Beteiligung und/oder Entwicklung einer pulmonalen Hypertonie
Zwischen behandelten und unbehandelten Patienten werden im Langzeitverlauf nur geringe Unterschiede beobachtet, und Rezidive treten nach Therapieende häufig auf.
Wichtige Punkte
Systemische und extrapulmonale Beteiligung ist häufig bei Sarkoidose, aber > 90% der erwachsenen Patienten weisen eine Lungenbeteiligung auf.
Eine bildgebende Untersuchung der Brust sollte durchgeführt, aber die Diagnose durch Biopsie bestätigt werden, in der Regel durch endobronchiale Ultraschall-geführte transbronchiale Adelpunktion eines mediastinalen oder hilaren Lymphknotens.
Der pulmonale Schweregrad sollte mit Lungenfunktionstests bewertet und eine Pulsoxymetrie durchgeführt werden.
Mittels EKG, Spaltlampenuntersuchung, Nieren- und Leberfunktionstests sowie Serum -und Urinkalziumtests sollte die extrapulmonale Beteiligung geprüft werden.
Behandlung von Patienten mit systemischen Kortikosteroiden, wenn angezeigt (z. B. schwere Symptome, Hyperkalzämie, progressive Rückgang der Organfunktion, Herz-oder neurologische Beteiligung).
Behandelung mit Immunsuppressiva, wenn Patienten moderate Dosen von Kortikosteroiden nicht tolerieren, die Sarkoidose resistent gegen Kortikosteroide ist oder wenn Kortikosteroide langfristig erforderlich sind.