




Primäre maligne Knochentumoren sind viel seltener als metastasierende Knochentumoren, insbesondere bei Erwachsenen. Zu den primären maligne Knochentumoren gehören multiples Myelom, Osteosarkom, Adamantinom, Chondrosarkom, Chordom, Ewing-Knochensarkom, Fibrosarkom und undifferenziertes pleomorphes Sarkom, Lymphom des Knochens, und bösartiger Riesenzelltumor. (Siehe auch Übersicht von Knochen- and Gelenktumoren und Übersicht über Leukämie.)
Die beiden am häufigsten verwendeten Systeme zur Stadieneinteilung dieser Tumoren sind:
The American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 8. Auflage: Bei Osteosarkomen, Chondrosarkomen und Ewing-Sarkomen basiert die Stadieneinteilung auf einer bestimmten Tumorkategorie, dem histologischen Grad, der Größe, der Nodalbeteiligung und den Metastasen (TNM-Klassifikation). Das Handbuch klassifiziert Tumoren in 4 Stadien und wird für die Meldung von Krebsdaten verwendet.
Das Staging-System der Musculoskeletal Tumor Society (MSTS): Wird von orthopädischen Onkologiechirurgen auf der Grundlage des histologischen Grades verwendet (z. B. Stadium I – niedriggradige Histologie und Stadium II – hochgradige Histologie, ob der Tumor vollständig innerhalb des Knochens liegt (A) oder aus dem Kortex in das umgebende Weichgewebe eingedrungen ist (B), und Metastasen Stadium III). Das typische (konventionelle) Osteosarkom mit einer assoziierten Weichteilmasse ohne Metastasen wird im MSTS-System dem Stadium IIB zugeordnet.
Multiples Myelom
Das Multiple Myelom ist der häufigste primäre bösartige Knochentumor, wird aber wegen seiner hämatopoetischen Abstammung oft als Markzelltumor innerhalb des Knochens und nicht als primärer Knochentumor angesehen (siehe auch Multiple Myeloma). Auch wenn das Multiple Myelom als hämatologischer Tumor gilt, muss die festgestellte Skelettanomalie von anderen Knochentumoren unterschieden werden.
Das multiple Myelom tritt meist bei älteren Erwachsenen auf.
Die Tumorentwicklung und -progression ist üblicherweise multizentrisch und befällt das Knochenmark so diffus, dass eine Knochenmarkpunktion diagnostisch beweisend ist. Anders als bei metastasierten Erkrankungen kann ein Radionuklid-Knochenscan Läsionen nicht zuverlässig zeigen, und daher sollten Skelettuntersuchungen durchgeführt werden. Skelettuntersuchungen zeigen typischerweise scharf begrenzte lytische Läsionen (gestanzte Läsionen) oder diffuse Demineralisation. Selten kann die Läsion als sklerotisch oder als diffuse Osteopenie erscheinen, vor allem in einem Wirbelkörper. Eine isolierte einzelne Myelomläsion ohne systemische Beteiligung des Knochenmarks wird als Plasmozytom bezeichnet.

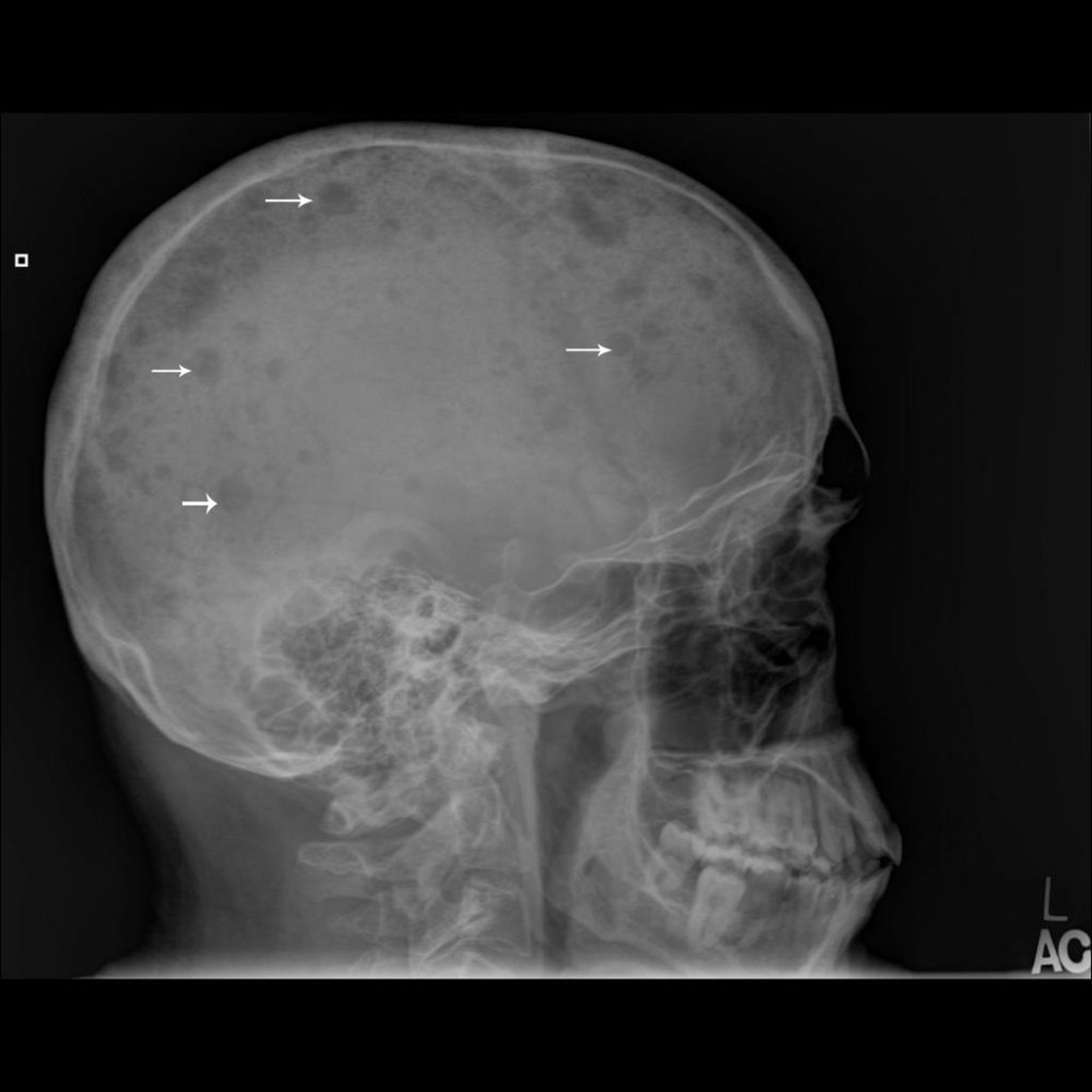
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Bestimmte Knochenläsionen reagieren sehr gut auf Strahlentherapie.
Osteosarkom (osteogenes Sarkom)
Das Osteosarkom ist der häufigste bösartige primäre Knochentumor (wenn man das Myelom als Markzelltumor und nicht als primären Knochentumor betrachtet) und ist hochmaligne. Das Osteosarkom kann in jedem Alter auftreten, am häufigsten jedoch zwischen dem 10. und 25. Lebensjahr. Es gibt zwei Inzidenzspitzen: Die höchste Inzidenz ist bei Jugendlichen und sehr jungen Erwachsenen zu verzeichnen (zeitgleich mit dem Wachstumsschub in der Pubertät), die zweite Spitze tritt bei älteren Erwachsenen (≥ 60 Jahre) auf, insbesondere bei Personen mit Risikofaktoren wie Paget-Krankheit, Knocheninfarkten und Knochenbereichen, die bereits viele Jahre zuvor wegen einer anderen Krebserkrankung einer hochdosierten Strahlentherapie ausgesetzt waren. Es besteht eine genetische Veranlagung, insbesondere bei Kindern, die Träger des Gens für das hereditäre Retinoblastom (Varianten des RB1-Gens) und des Li-Fraumeni-Syndroms (TP53-Gen) sind.
Es produziert malignes Osteoid (unreifen Knochen) aus tumorösen Knochenzellen. Üblicherweise ist es um das Knie herum (das distale Femur ist häufiger betroffen als die proximale Tibia) oder in anderen Röhrenknochen, speziell in den metaphysär-diaphysären Arealen, zu finden. Metastasen zeigen sich am häufigsten in Lunge oder am Knochen, Schmerz und Schwellung sind die häufigsten Symptome.

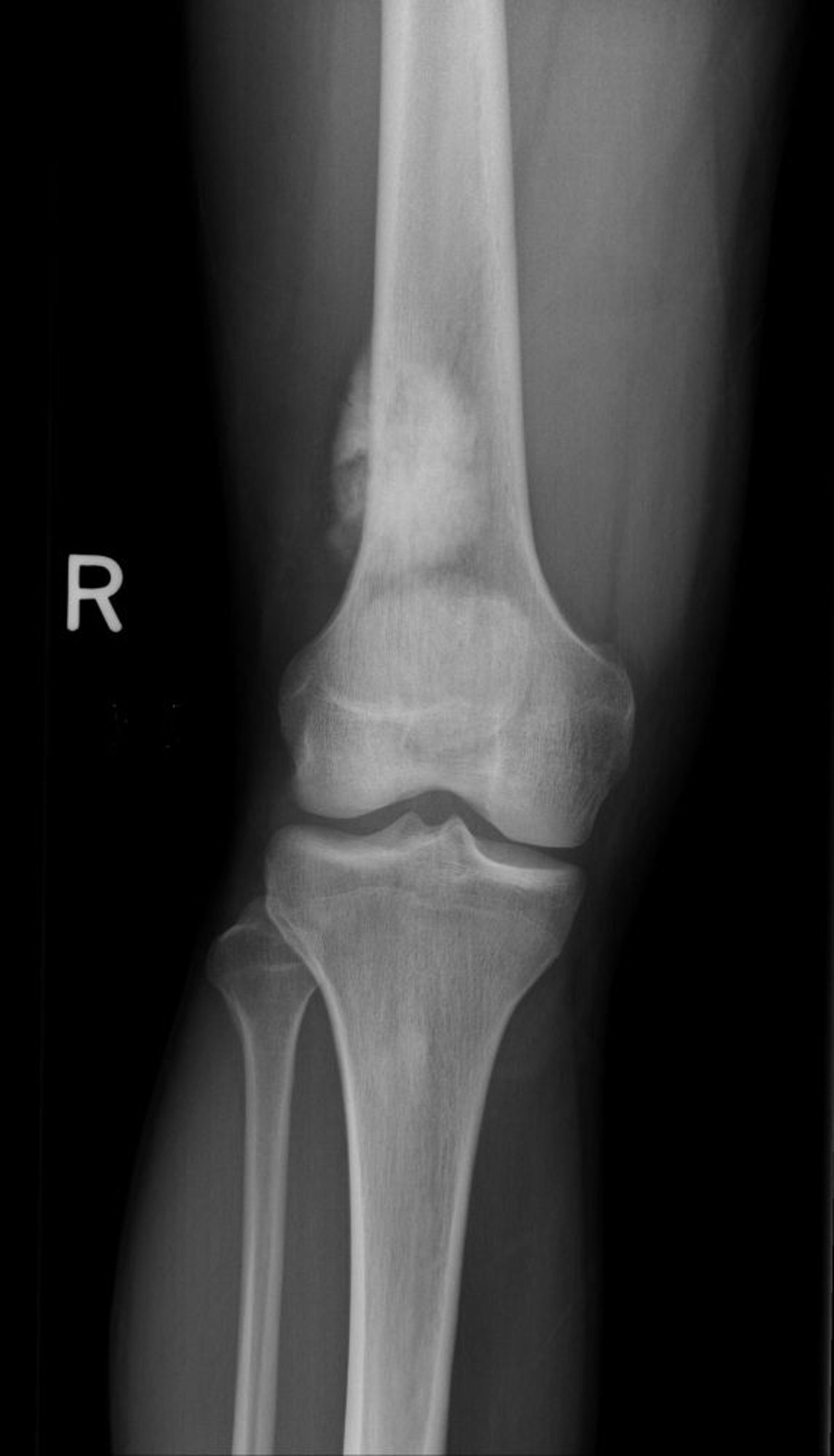
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Die Befunde bildgebender Untersuchungen variieren, sklerotische und lytische Muster kommen vor. Die Diagnose eines Osteosarkoms erfordert eine Biopsie. mittels Röntgenthorax und CT wird nach Lungenmetastasen gesucht, mittels Skelettszintigraphie nach Knochenmetastasen. Eine MRT der gesamten betroffenen Extremität wird durchgeführt, um - falls vorhanden - metachrone Läsionen zu detektieren. Eine PET-CT kann Fernmetastasen oder metachrone Läsionen zeigen.
Die Behandlung des Osteosarkom besteht in einer Kombination aus Chemotherapie und chirurgischer Intervention. Der Einsatz adjuvanter Chemotherapie erhöht die Überlebensrate von < 20% auf > 65% in 5 Jahren. Die neoadjuvante Chemotherapie beginnt vor der chirurgischen Resektion. Eine verringerte periphere Weichteiltumormasse oder eine erhöhte Mineralisierung auf Röntgenbildern, eine verringerte Schmerzintensität und eine verringerte alkalische Phosphatase im Serum deuten auf ein gewisses Ansprechen hin, aber das gewünschte Ansprechen ist eine Tumornekrose von > 95% bei der histologischen Darstellung des resezierten Präparats durch den Pathologen. Nach mehreren Chemotherapiezyklen (über mehrere Monate) kann eine gliedmaßenerhaltende Operation und eine Rekonstruktion der Gliedmaße durchgeführt werden. Gelegentlich wird eine chirurgische Amputation vor Beginn der Chemotherapie bei einem verpilzenden Tumor durchgeführt. Ziel ist es, die frühe mikrometastatische Erkrankung zu behandeln, von der angenommen wird, dass sie vorhanden ist, auch wenn sie in den bildgebenden Untersuchungen zum Staging nicht gesehen wird.
Bei der gliemaßenerhaltenden Operation wird der Tumor en bloc reseziert, einschließlich des gesamten umgebenden reaktiven Gewebes und einem Rand des umgebenden gesunden Gewebes. Um ein mikroskopisches Streuen von Tumorzellen zu vermeiden, darf der Tumor nicht verletzt werden. Mehr als 85% der Patienten können mit einer gliedmaßenerhaltenden Operation behandelt werden, ohne dass sich die langfristige Überlebensrate verringert.
Die Fortführung der Chemotherapie nach der Operation ist notwendig. Wenn es durch die präoperative Chemotherapie zu einer fast vollständigen Tumornekrose (ca. 95%) kommt, beträgt die 5-Jahres-Überlebensrate > 90%. Begrenzte Metastasen in der Lunge können manchmal durch Thorakotomie mit Keilresektion der Lungenläsion behandelt werden.
Zu den Varianten des Osteosarkoms, die sich vom herkömmlichen Osteosarkom unterscheiden und viel seltener auftreten, gehören oberflächliche Kortikalisläsionen wie das parosteale Osteosarkom und das periosteale Osteosarkom. Parossale Osteosarkome betreffen meist den posterioren Kortex des distalen Femurs und sind in der Regel relativ gut differenziert. Bei der Behandlung von niedriggradigen parostealen Osteosarkomen ist eine Chemotherapie vor der chirurgischen Resektion nicht erforderlich. Parosteale Osteosarkome erfordern eine chirurgische En-bloc-Resektion, aber keine Chemotherapie, wenn die Histologie des resezierten Präparats bestätigt, dass der Tumor gut differenziert ist.
Das periostale Osteosarkom ist eher ein Knorpelmatrix-Oberflächentumor, der auch Knochenmatrix enthält und maligne ist. Es ist oft auf der Schaftmitte des Femurs lokalisiert und stellt sich im Röntgenbild als Sonnenausbruch dar. Die Wahrscheinlichkeit von Metastasen beim periostalen Osteosarkom ist viel größer als beim gut differenzierten parossalen Osteosarkom, aber etwas geringer als beim typischen Osteosarkom. Zumeist werden periostale Osteosarkome ähnlich wie konventionelle Osteosarkome mit Chemotherapie und chirurgischer En-bloc-Resektion behandelt.
Adamantinom
Das Adamantinom ist selten (< 1% der malignen Knochentumoren) und tritt am häufigsten in der Tibia auf. Sie tritt in der Regel bei Heranwachsenden und Menschen in der 3. Lebensdekade auf, kann aber grundsätzlich in jedem Alter auftreten. Das Adamantinom wächst langsam und manifestiert sich oft mit Schmerzen und tastbarer Füllung.

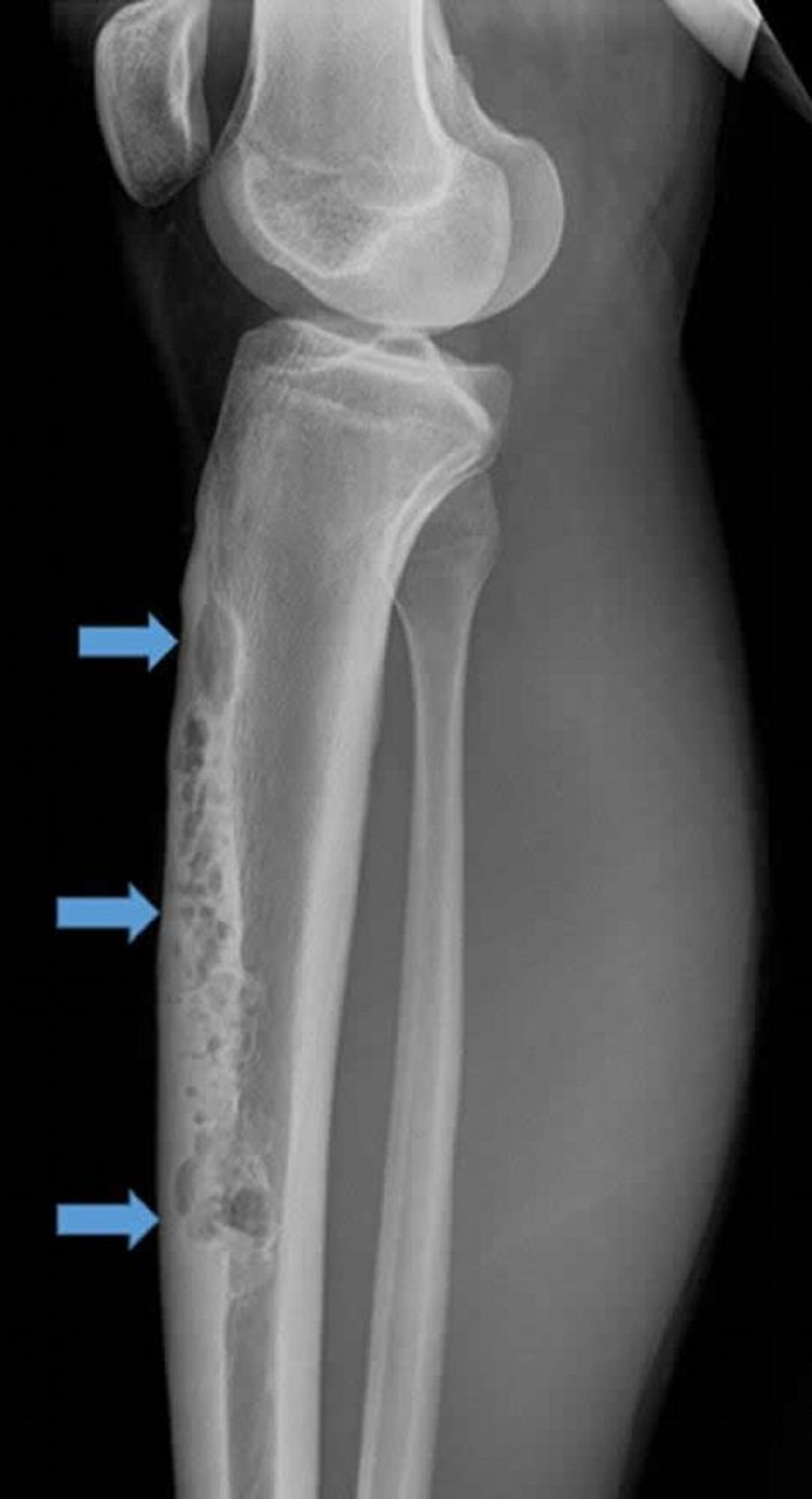
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Die Läsion manifestiert sich typischerweise in der Tibiavorderkante, und die Röntgenbilder zeigen das osteolytische Erscheinungsbild einer "Seifenblase". Das histologische Erscheinungsbild ist ein zweiphasiges Muster aus epithelialem und osteofibrösem Gewebe. Die Läsion kann mit einer osteofibrösen Dysplasie der vorderen Tibiakortikalis verwechselt werden, die gutartig ist. Einige Kliniker glauben, dass die osteofibröse Dysplasie der vorderen Tibiakortikalis ein Vorläufer des Adamantinoms sein könnte, jedoch ohne die epitheliale Komponente, die es zu einem Tumor machen würde.
Metastasen treten vor allem in der Lunge auf, sind aber selten.
Die Behandlung des Adamantinoms besteht aus einer breiten Resektion und Rekonstruktion des Defekts. Gelegentlich ist eine Amputation notwendig.
Chondrosarkom
Chondrosarkome sind maligne Tumoren des Knorpels. Sie unterscheiden sich von Osteosarkomen klinisch, therapeutisch und prognostisch. 90% der Chondrosarkome sind primär. Chondrosarkome können auch bei anderen Vorerkrankungen entstehen, insbesondere bei multiplen Osteochondromen und bei multipler Enchondromatose (z. B. bei der Ollier-Krankheit und beim Maffucci-Syndrom). Chondrosarkome kommen bevorzugt bei älteren Erwachsenen vor. Sie entwickeln sich häufig in flachen Knochen (z. B. Becken, Schulterblatt), können aber in jedem Teil eines beliebigen Knochens entstehen (am häufigsten im Oberschenkelknochen und im Oberarmknochen unter den langen Knochen) und können eine Weichteiltumorkomponente haben, die das umgebende Weichgewebe mit einbezieht.

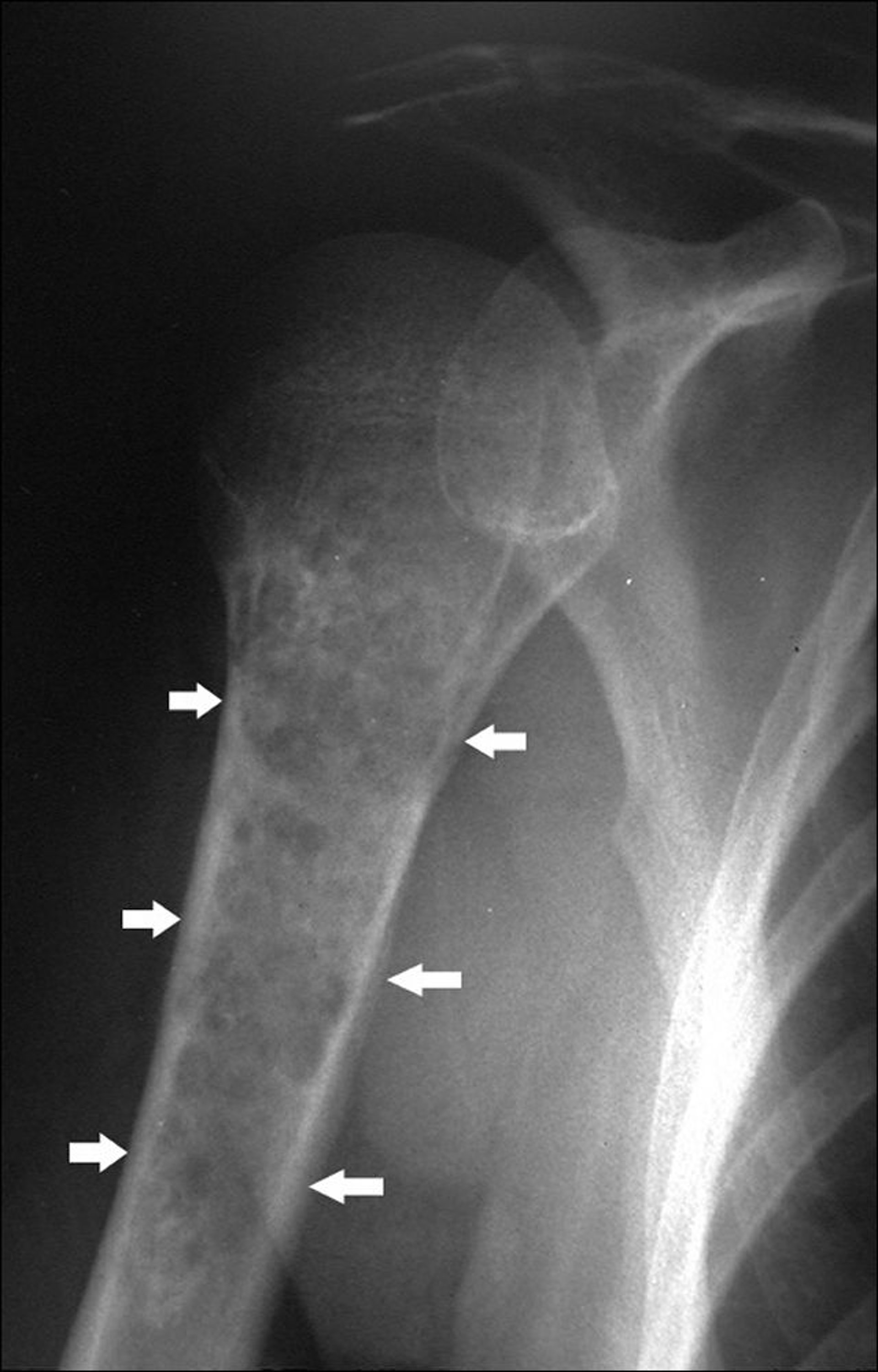
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Röntgenaufnahmen zeigen häufig punktuelle Kalzifikationen. Chondrosarkome bieten ebenso häufig eine Zerstörung des kortikalen Knochens und den Verlust von normaler trabekulärer Knochenstruktur. Die MRT kann eine Weichteilmasse aufweisen. Es kann auch ein Knochenscan durchgeführt werden. Für Chondrosarkom ist die histologischen Diagnose erforderlich, die auch den Grad des Tumors festlegen kann (v. a. der Metastasierung). Die Nadelbiopsie kann eine unzureichende Gewebeprobe liefern.
Es ist oft schwierig, niedriggradige Chondrosarkome von Enchondromen durch Bildgebung und manchmal sogar Histologie zu unterscheiden.
Niedriggradige Chondrosarkome (Grad 1) werden oft intraläsional (weite Kürettage) unter Zugabe eines Adjuvans (oft Einfrieren mit flüssigem Stickstoff, Argonbeamer, Wärme aus Methylmethacrylat, Radiofrequenz oder Phenol) behandelt. Einige Chirurgen bevorzugen eine chirurgische En-bloc-Resektion für Tumore niedriger Qualität, um das Rezidivrisiko zu verringern. Die Diagnose eines Osteosarkoms erfordert eine Biopsie. Wenn die chirurgische Resektion mit Aufrechterhaltung der Funktion nicht möglich ist, kann eine Amputation notwendig werden. Wegen Metastasierungsgefahr muss höchste Sorgfalt bei der Durchführung einer Biopsie oder Operation herrschen, um ein Streuen von Tumorzellen in das Weichgewebe zu vermeiden. Sollte der Tumor streuen, ist eine Heilung unerreichbar. Wenn es nicht zu einer Streuung kommt, ist die Heilungsrate vom Tumorgrad abhängig. Niedriggradige Tumoren haben eine fast vollständige Heilungsrate bei adäquater Therapie. Da diese Tumoren eine eingeschränkte Durchblutung aufweisen, zeigen Chemo- und Strahlentherapie wenig Wirksamkeit.
Chordom
Das Chordom, das selten vorkommt, entwickelt sich aus den Überresten des primitiven Notochords. Die Prädilektionsstellen liegen an den Enden der Wirbelsäule, gewöhnlich in der Mitte des Os sacrum oder nahe der Schädelbasis. Ein Chordom in der sakrokokzygealen Region verursacht einen konstanten Schmerz. Ein Chordom an der Schädelbasis kann ein Defizit eines Hirnnervs verursachen, meist sind die augenversorgenden Nerven betroffen.
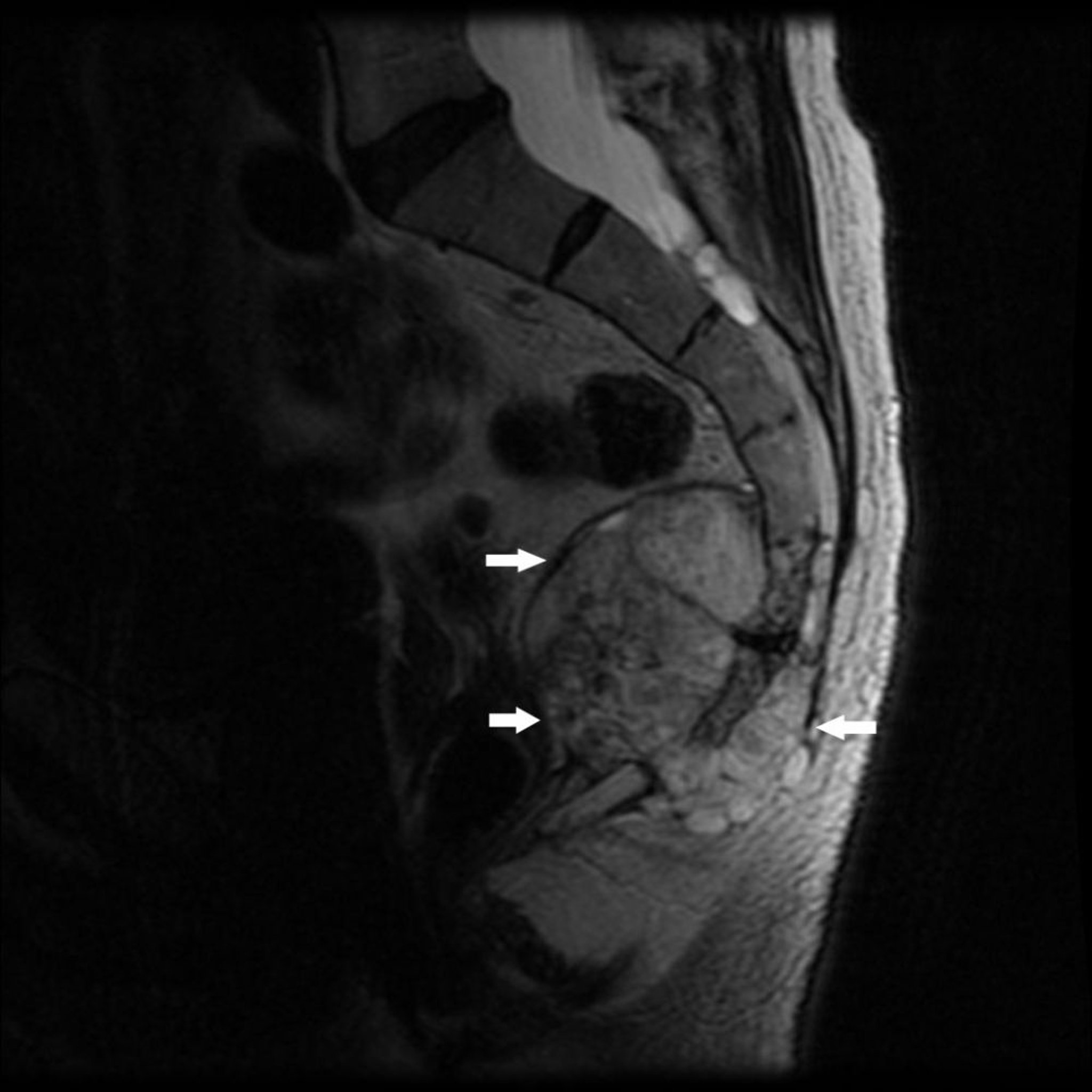
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Die Symptome eines Chordoms können mehrere Monate bis Jahre der Diagnose vorausgehen.
Ein Chordom erscheint in bildgebenden Untersuchungen als eine destruktive Knochenläsion, die mit einer Weichteilmasse assoziiert sein kann. Eine Biopsie wird durchgeführt.
Chordome in der sakrokokzygealen Region können durch eine radikale En-bloc-Exzision geheilt werden. Chordome an der Schädelbasis sind gewöhnlich operativ unzugänglich, können aber auf eine Bestrahlung ansprechen. Die Rate an Lokalrezidiven ist nach einer Tumorresektion hoch. Metastasen können, obwohl weniger häufig, auftreten.
Ewing-Sarkom des Knochens
Das Ewing-Sarkom des Knochens ist ein kleiner, blauer, rundzelliger Knochentumor mit einem Häufigkeitsgipfel zwischen 10 und 20 Jahren. Das Ewing-Sarkom ist mit den peripheren primitiven neuroektodermalen Tumoren (PNET) und dem malignen kleinzelligen Askin-Tumor der Brustwand verwandt, die heute zur Familie der Ewing-Sarkome gezählt werden. Die meisten Tumoren entwickeln sich in den Extremitäten, allerdings kann jeder Knochen betroffen sein. Das Ewing-Sarkom neigt zu extensivem Wachstum und befällt manchmal den gesamten Knochenschaft, am häufigsten die diaphysäre Region. Etwa 15–20% der Fälle betreffen die metaphysäre Region. Schmerz und Schwellung sind die häufigsten Symptome.

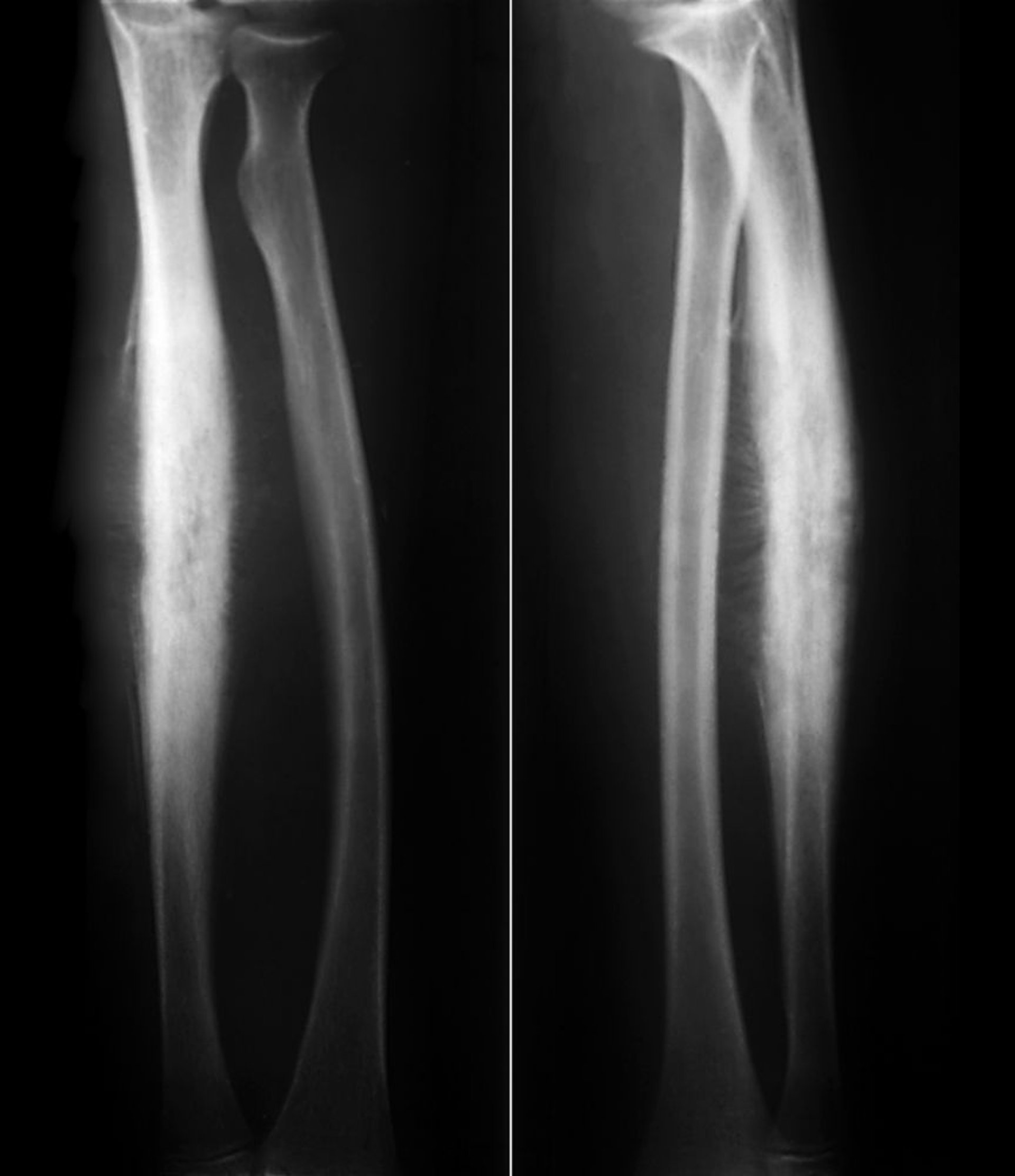
ZEPHYR/SCIENCE PHOTO LIBRARY
Eine lytische Destruktion, insbesondere ein permeatives Infiltrationsmuster ohne klare Begrenzung, ist der häufigste Befund bei bildgebenden Untersuchungen, es können jedoch auch multiple Schichten subperiostaler reaktiver Knochenneubildung einen zwiebelschalenartigen Aspekt verursachen. Röntgenaufnahmen offenbaren in der Regel nicht das ganze Ausmaß der Knochenbeteiligung, und in der Regel umgibt eine große Weichteilmasse den betroffenen Knochen. Das MRT definiert das Krankheitsausmaß am besten, was bei der Therapieplanung helfen kann.
Viele weitere benigne oder maligne Tumoren können sehr ähnlich erscheinen, sodass die Diagnose des Ewing-Sarkoms bioptisch gestellt werden muss. Bisweilen wird diese Art von Tumor mit einer Infektion verwechselt. Eine genaue histologische Diagnose kann durch eine zytogenetische Analyse und die Identifizierung molekularer Marker, einschließlich der Untersuchung auf eine typische klonale Chromosomenanomalie, gestellt werden. Molekulare Befunde in der Ewing-Tumorfamilie sind verschiedene nicht zufällige chromosomale Translokationen, die das Ewing-Sarkom-Gen (EWS) auf Chromosom 22 betreffen. Es wurden achtzehn verschiedene strukturelle Translokationen in unterschiedlichen Fusionsgenmustern festgestellt.

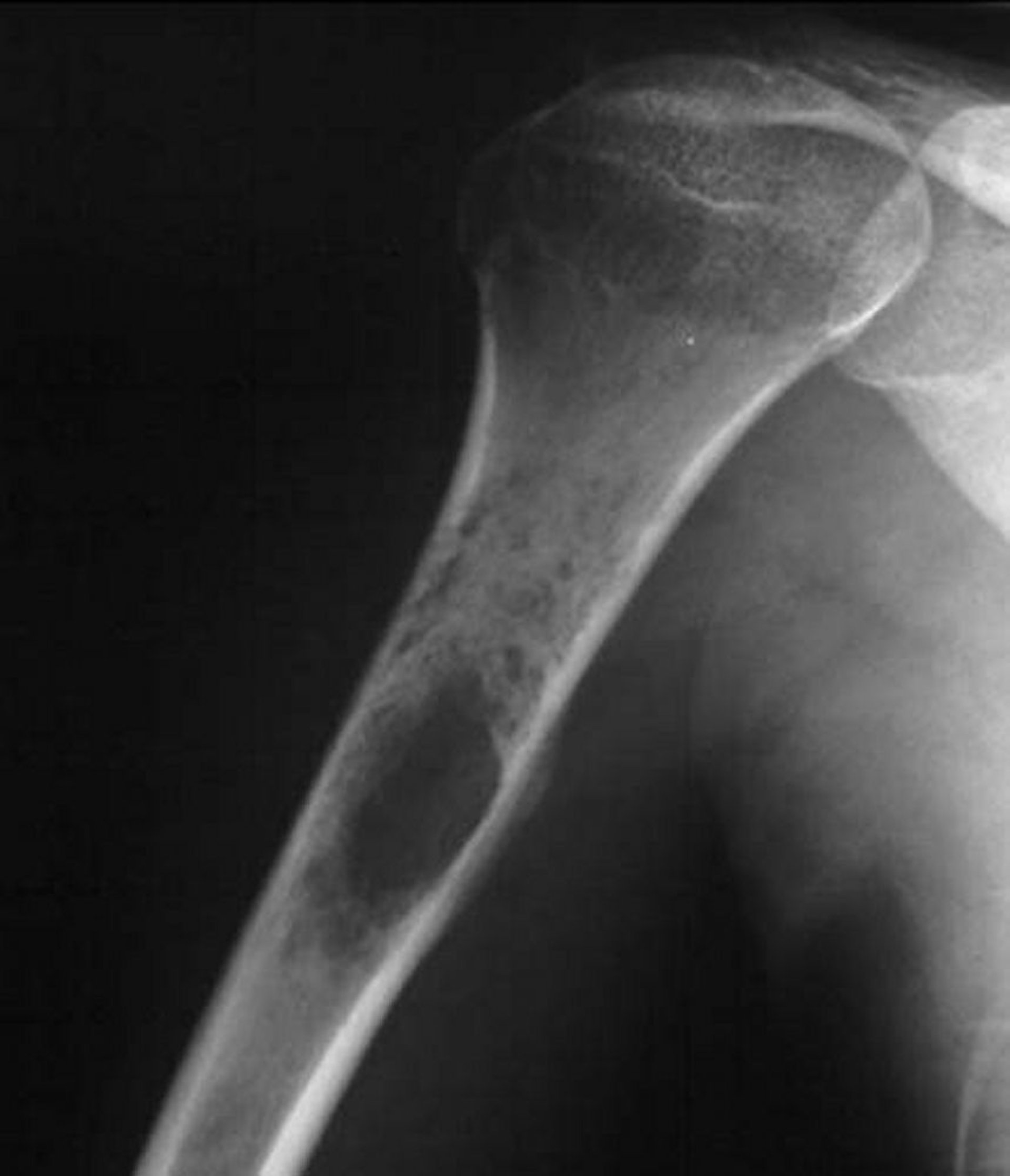
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Die Behandlung des Ewing-Sarkoms schließt verschiedene Kombinationen aus Chirurgie, Chemotherapie und Radiotherapie ein. Gegenwärtig können > 60% der Patienten mit primär lokalisiertem Ewing-Sarkom durch diese Therapieansätze geheilt werden. Eine Heilung ist gelegentlich sogar bei metastasierender Erkrankung möglich. Eine Chemotherapie in Verbindung mit einer chirurgischen En-Bloc-Resektion, falls zutreffend, führt häufig zu besseren Langzeitergebnissen als eine Chemotherapie in Verbindung mit einer Strahlentherapie.
Fibrosarkom und undifferenziertes pleomorphes Sarkom (früher malignes fibröses Histiozytom des Knochens)
Fibrosarkome und undifferenziertes pleomorphes Sarkom zeigen ähnliche Charakteristika wie Osteosarkome, produzieren jedoch fibröse Tumorzellen (statt Knochentumorzellen), betreffen dieselbe Altersgruppe und stellen die gleichen Probleme.
Behandlung und Outcome bei hochgradigen Läsionen sind ähnlich wie beim Osteosarkom.
Lymphom des Knochens
Das Knochenlymphom (früher bekannt als Retikulumzellsarkom) betrifft Erwachsene, gewöhnlich im 4. und 5. Lebensjahrzehnt. In der Regel handelt es sich um ein diffuses großzelliges B-Zell-Lymphom. Es kann in jedem Knochen entstehen. Der Tumor besteht aus kleinen, runden Zellen, häufig einer Mischung aus Retikulozyten, Lymphoblasten und Lymphozyten. Er kann als isolierter primärer Knochentumor oder in Assoziation mit ähnlichen Tumoren in anderen Geweben entstehen oder auch als Metastase bei bekanntem Weichteillymphom. Schmerzen und Schwellungen sind die üblichen Symptome eines Knochenlymphoms. Pathologische Frakturen häufig.

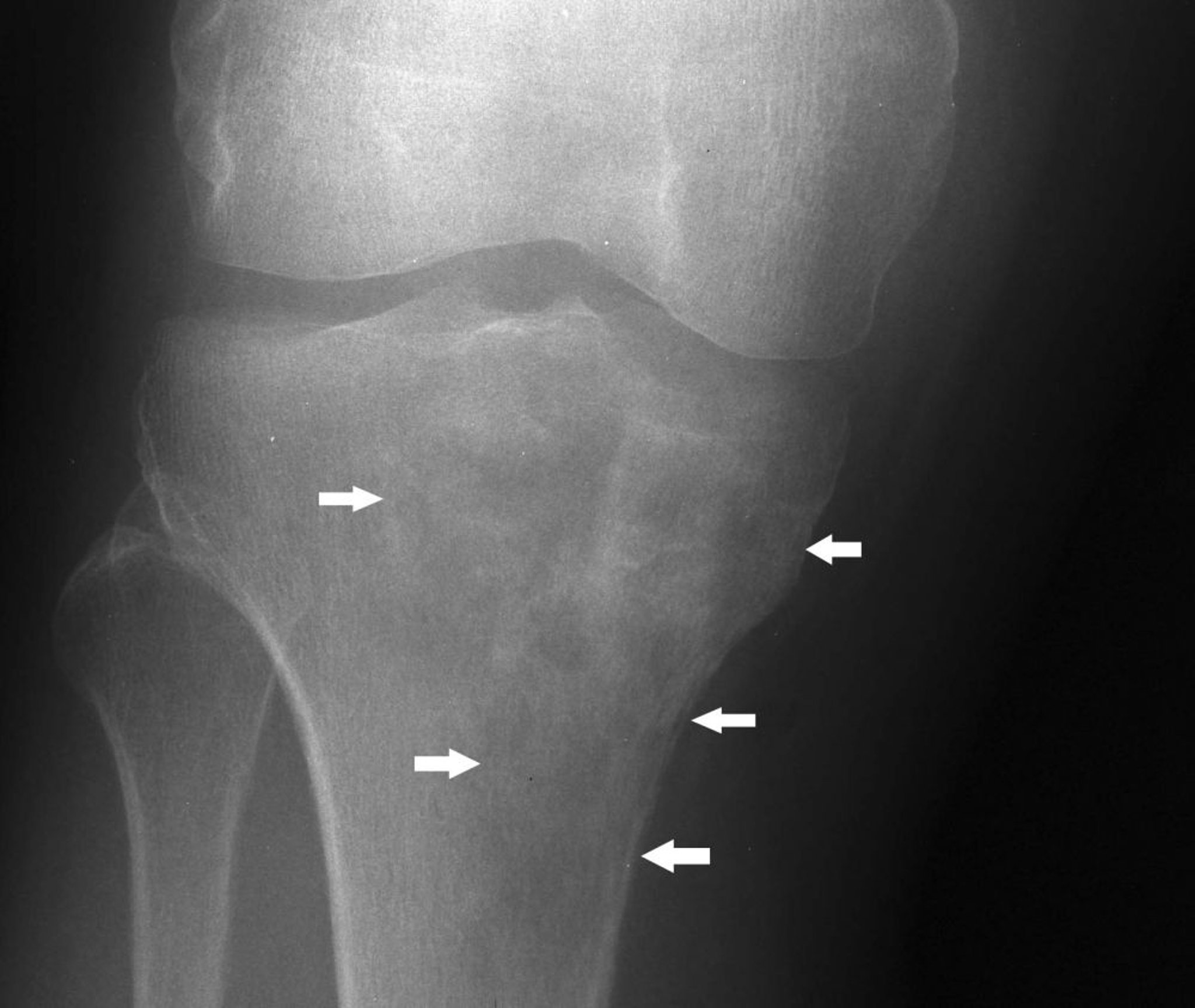
Image courtesy of Michael J. Joyce, MD, und Hakan Ilaslan, MD.
Bildgebende Untersuchungen zeigen eine Knochendestruktion, die mottenfraßähnlich oder fleckförmig oder sogar als infiltrierendes, permeatives Muster erscheint, oft mit einer klinischen und radiologischen großen Weichteilmasse. Bei fortgeschrittener Erkrankung kann der gesamte Umriss des betroffenen Knochens aufgelöst sein. Eine Biopsie wird ebenfalls durchgeführt.
Beim isolierten primären Knochenlymphom beträgt die 5-Jahres-Überlebensrate ≥ 50%.
Knochenlymphome werden typischerweise mit systemischer Chemotherapie behandelt. Die Strahlentherapie kann in einigen Fällen als Adjuvans eingesetzt werden. Eine Stabilisierung der langen Röhrenknochen ist oft notwendig, um pathologische Frakturen zu verhindern. Die Amputation ist nur selten indiziert, z. B. bei Funktionsverlust durch pathologische Frakturen oder wenn eine extensive Weichteilbeteiligung nicht auf andere Weise beseitigt werden kann.
Maligner Riesenzelltumor
Der sehr seltene maligne Riesenzelltumor ist gewöhnlich an den Extremitäten am Ende eines langen Röhrenknochens zu finden.
Röntgenbilder zeigen die klassischen Befunde einer malignen Destruktion (vorherrschende lytische Destruktion, kortikale Destruktion, Ausdehnung ins Weichteilgewebe und pathologische Frakturen). Ein maligner Riesenzelltumor, der sich aus einem vorbestehenden benignen Riesenzelltumor entwickelt hat, ist charakteristischerweise strahlenresistent. MRT und Biopsie werden durchgeführt.
Die Behandlung des bösartigen Riesenzelltumors ist ähnlich wie bei Osteosarkom, aber die Heilungsrate ist gering.