




Das bullöse Pemphigoid ist eine chronische Autoimmunerkrankung der Haut, die bei älteren Patienten zu generalisierten, juckenden, bullösen Läsionen führt. Die Schleimhaut ist selten betroffen. Die Diagnose erfolgt mittels Hautbiopsie und Immunfluoreszenztests von Haut und Serum. Zunächst werden topische und systemische Kortikosteroide verwendet. Bei den meisten Patienten ist eine Langzeit-Erhaltungstherapie, für die verschiedene Immunsuppressiva verwendet werden können, erforderlich.
Bullae sind erhöhte, mit Flüssigkeit gefüllte Blasen mit ≥ 10 mm Durchmesser.
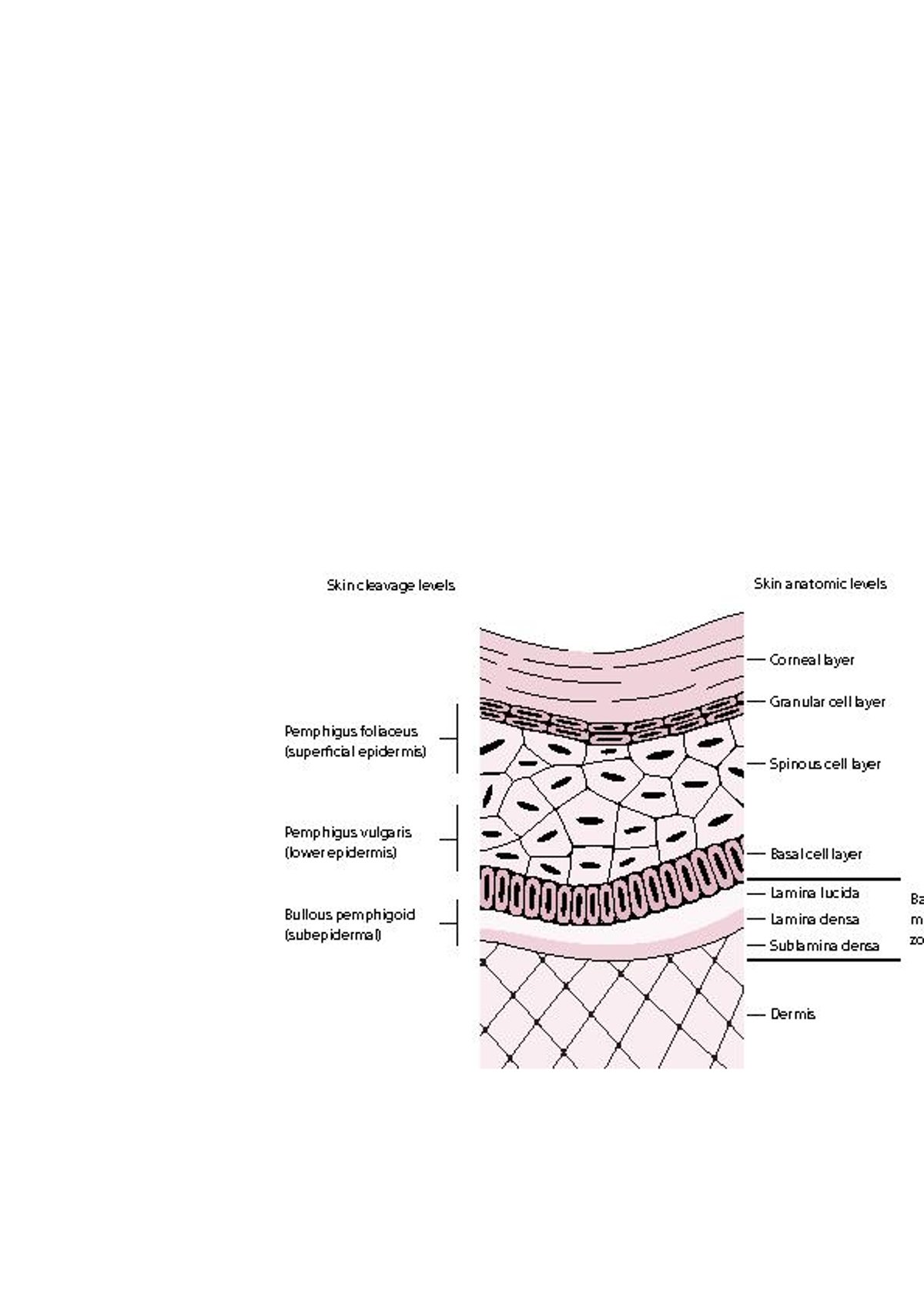
Das bullöse Pemphigoid tritt häufiger bei Patienten > 60 Jahren auf, kann aber auch bei Kindern vorkommen. IgG-Autoantikörper binden an bestimmte hemidesmosomale Antigene (BPAg1, [BP230], BPAg2, [BP180]). Dadurch wird eine Komplementaktivierung ausgelöst, die wiederum zur Bildung einer subepidermalen Blase führt (siehe Abbildung Grad der Hautschädigung bei Pemphigus und bullösem Pemphigoid).
Grad der Hautschädigung bei Pemphigus und bullösem Pemphigoid
Beim Pemphigus foliaceus bilden sich Bläschen in den oberflächlichen Schichten der Epidermis. Pemphigus-vulgaris-Bläschen können sich auf jeder epidermalen Ebene bilden, bilden sich aber typischerweise in den unteren Bereichen der Epidermis. Beim bullösen Pemphigoid bilden sich die Blasen subepidermal (Lamina lucida der Basalmembranzone). In dieser Abbildung ist die Basalmembranzone zur Darstellung ihrer Schichten überproportional vergrößert. |

Ätiologie des bullösen Pemphigoid
Es wurde keine Ursache für das bullöse Pemphigoid nachgewiesen, jedoch wurden die folgenden Auslöser vorgeschlagen:
Arzneimittel (einschließlich Furosemid, Spironolacton, Omeprazol, PD-1 und PD-L1 monoklonale Antikörper [z. B. Durvalumab, Nivolumab, Pembrolizumab], Sulfasalazin, Penicillin, Penicillamin, Etanercept, Antipsychotika und Dipeptidyl-Peptidase-4-Inhibitoren)
Körperliche Auslöser (einschließlich Trauma, Strahlentherapie für Brustkrebs, UV-Strahlung und Dithranol (Cignolin)
Erkrankungen der Haut (einschließlich Psoriasis, Lichen ruber, und einige Infektionen)
Störungen (Diabetes mellitus, rheumatoide Arthritis, Colitis ulcerosa und multiple Sklerose)
Genetische- und Umweltfaktoren können eine Rolle spielen.
Auslöser können eine Autoimmunreaktion durch Nachahmen von Molekülsequenzen in der epidermalen Basalmembran (molekulares Mimikry, wie bei Arzneimitteln und möglicherweise Infektionen) induzieren, durch Exposition oder Umwandlung normalerweise tolerierter Wirtsantigene (wie bei physikalischen Auslösern und bestimmte Erkrankungen) oder durch andere Mechanismen. Die Ausbreitung von Epitopen bezieht sich auf die Rekrutierung von autoreaktiven Lymphozyten gegen normalerweise tolerierte Wirtsantigene, die zur Chronifizierung der Krankheit und Verlauf beiträgt.
Bestimmte Erkrankungen des Zentralnervensystems (ZNS) und psychiatrische Störungen können dem bullösen Pemphigoid vorausgehen, insbesondere Multiple Sklerose und Schizophrenie, aber auch Demenzen, intrakranielle Blutungen, Schlaganfall, Wahn- und Persönlichkeitsstörungen sowie die Parkinson-Krankheit. In geringerem Maße kann diesen Störungen ein bullöses Pemphigoid vorausgehen. Zu den vermuteten gemeinsamen Ursachen gehören eine kreuzreaktive Immunantwort zwischen neuralen und kutanen Antigenen (BPAg1 wird im ZNS exprimiert) sowie die Auslösung durch bestimmte Medikamente, die zur Behandlung der Erkrankungen eingesetzt werden (z. B. Phenothiazin-Antipsychotika, Spironolacton); ein Mechanismus der Auslösung durch Medikamente ist jedoch nicht verstanden.
Symptome und Anzeichen von bullösem Pemphigoid
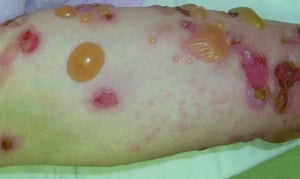
Pruritus ist das erste Symptom des bullösen Pemphigoid. Hautläsionen können sich über mehrere Jahre nicht entwickeln. Häufig entwickeln sich auf der Haut des Rumpfes und in den Biege- und Intertriginalbereichen charakteristische Spannungsgefühle. Bullae kann sich auf normal erscheinender Haut entwickeln oder erythematösen oder urtikariösen Plaques vorangehen. Lokalisierte Krankheit kann an Stellen des Traumas, der Stomata und in den anogenitalen und Unterschenkelbereichen auftreten. Das dyshidrotische Pemphigoid ist eine seltene Form des bullösen Pemphigoids, die Hände und Füße betrifft und an den Handflächen wie eine dyshidrotische Dermatitis (eine Form der Hand- und Fußdermatitis) aussehen kann. Blasen platzen in der Regel nicht auf. Wenn sie es doch tun, heilen sie oft schnell.
Zusätzlich können polymorphe, anuläre, dunkelrote, ödematöse Läsionen mit peripheren Bläschen oder ohne auftreten. Selten entwickeln sich kleine Bläschen auf der Schleimhaut. Leukozytose und Eosinophilie sind häufig, aber Fieber ist selten. Das Nikolski-Phänomen, bei dem sich die oberen Schichten der Epidermis bei leichtem Druck oder dem Reiben der Haut direkt neben einer Blase lateral bewegen, ist negativ.
© Springer Science+Business Media
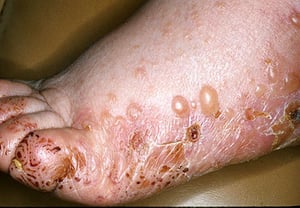
Image provided by Thomas Habif, MD.
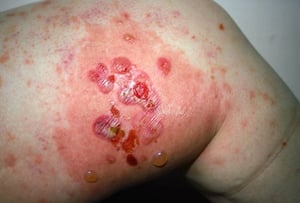
Image courtesy of Karen McKoy, MD.
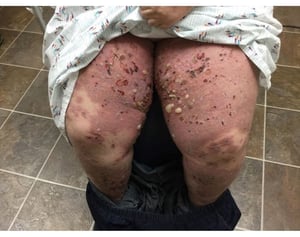
Image courtesy of Daniel M. Peraza, MD.

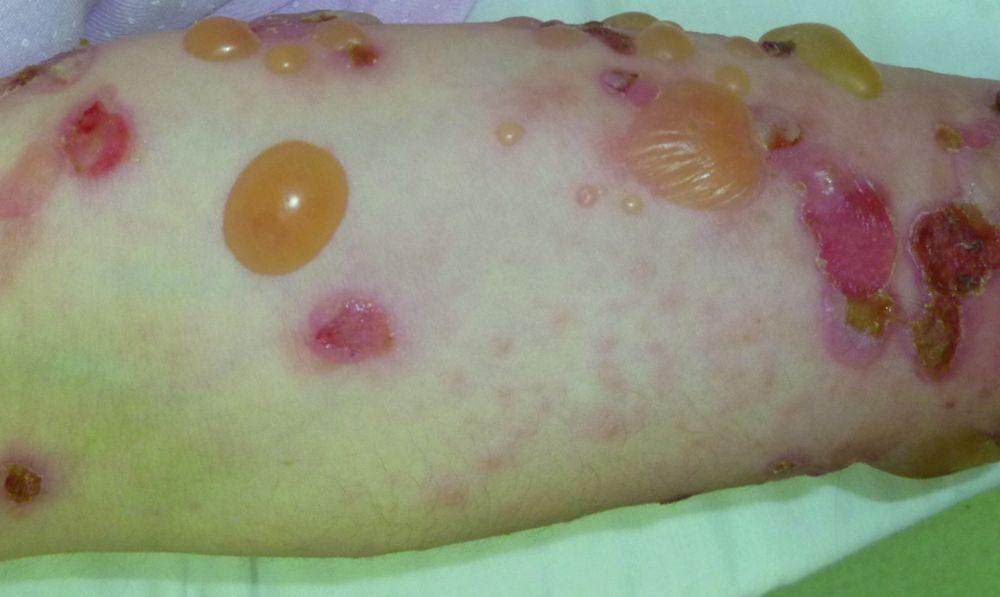
© Springer Science+Business Media

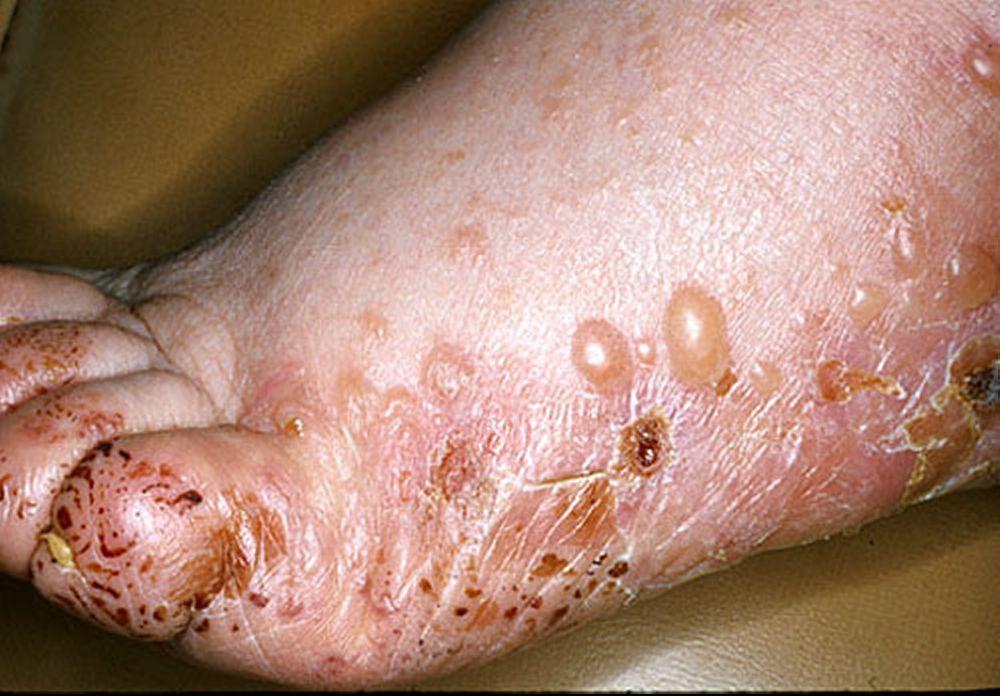
Image provided by Thomas Habif, MD.

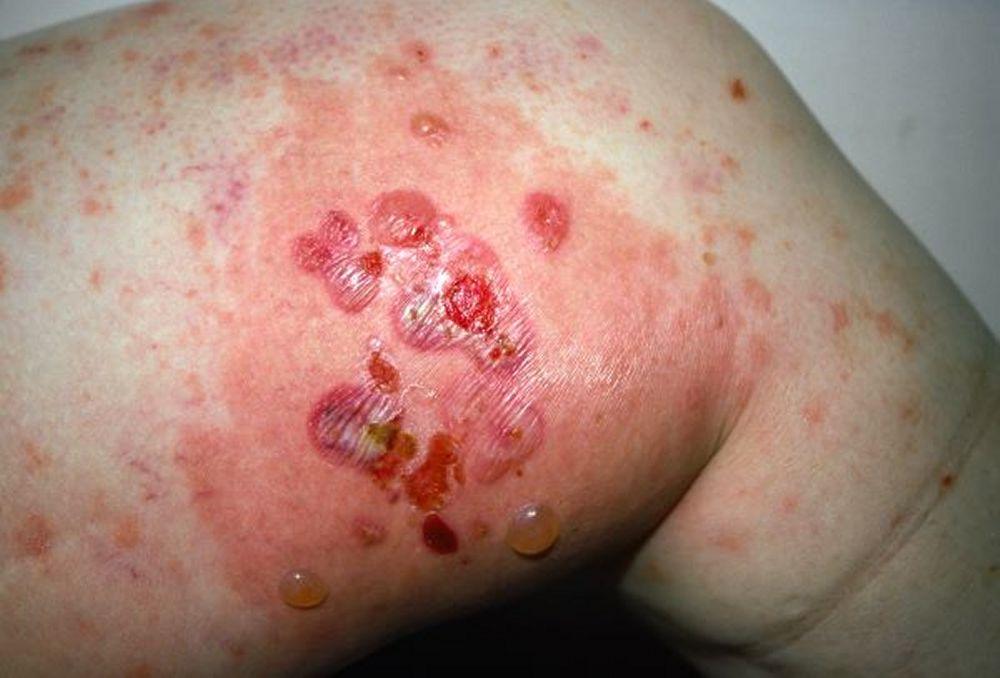
Image courtesy of Karen McKoy, MD.

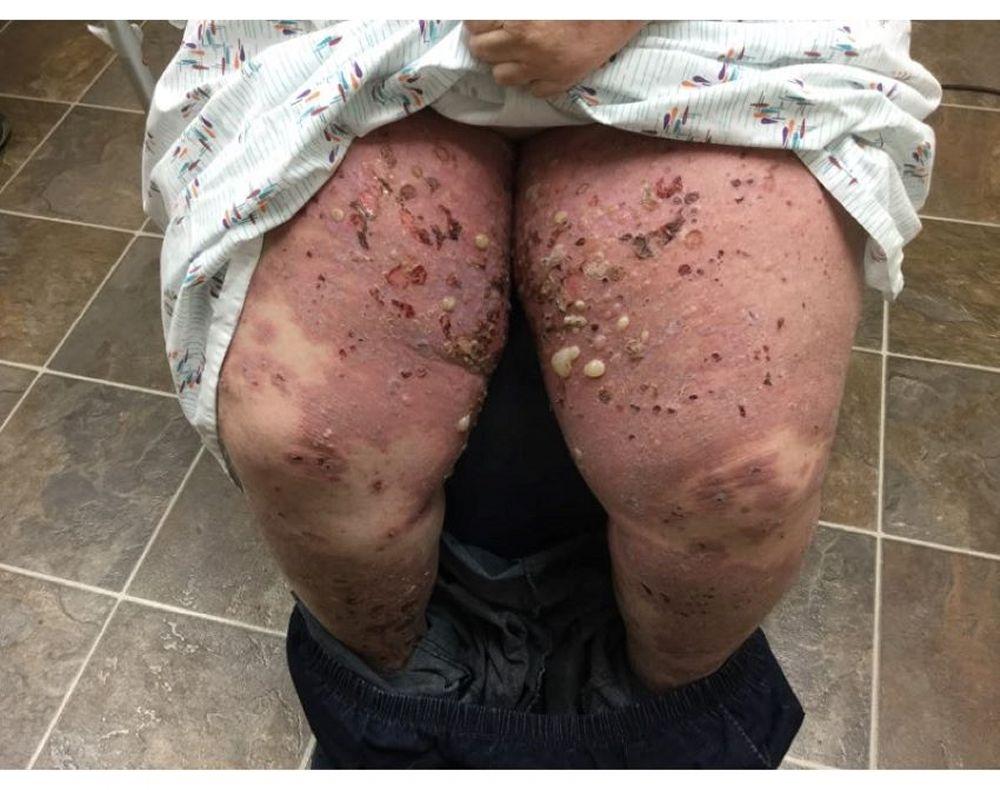
Image courtesy of Daniel M. Peraza, MD.
Diagnose von bullösem Pemphigoid
Hautbiopsie und IgG-Titer
Wenn sich Blasen entwickeln, muss das bullöse Pemphigoid vom Pemphigus vulgaris, einer blasenbildenden Erkrankung mit einer schlechteren Prognose, differenziert werden. Die Differenzierung ist in der Regel anhand von klinischen Kriterien ( siehe Tabelle: Unterscheidung zwischen bullösem Pemphigoid und Pemphigus vulgaris) möglich.
Testergebnisse helfen dabei, das bullöse Pemphigoid von dem Pemphigus vulgaris, der linearen IgA-Dermatose, dem Erythema (exsudativum) multiforme, Arzneimittelexanthemen, dem Schleimhautpemphigoid, dem paraneoplastischen Pemphigoid, der Dermatitis herpetiformis Duhring und der Epidermolysis bullosa acquisita abzugrenzen.
Wenn der Verdacht auf ein bullöses Pemphigoid besteht, werden eine Hautbiopsie für die Histologie und die direkte Immunfluoreszenz durchgeführt. Proben von der Haut der Läsion selbst und von der Haut um die Läsion herum werden oft für die Histologie genutzt, aber für die direkte Immunfluoreszenz werden Proben von unbetroffener Haut (oft etwa 3 mm vom Rand einer Läsion entfernt) verwendet. Die Blase ist beim bullösen Pemphigoid subepidermal, oft mit vielen Neutrophilen und Eosinophilen. Direkte Immunfluoreszenz zeigt lineare IgG- und Komplementablagerungen entlang der Basalmembranzone (dermal-epidermaler Übergang). Indirekte Immunfluoreszenz zeigt zirkulierende IgG-Ablagerungen auf der epidermalen Seite eines Salzspaltpräparats normaler Haut (d. h. Testsubstrat).
Das Serum wird mit Hilfe des ELISA (enzyme-linked immunosorbent assay) auf IgG-Antikörper gegen BPAg1 und BPAg2 getestet. Zirkulierende IgG-Autoantikörper sind bei etwa drei Viertel der Patienten vorhanden.
Prognose für bullöses Pemphigoid
Bullöses Pemphigoid ist eine chronische, potenziell tödliche Krankheit, insbesondere ohne Behandlung. Obwohl topische und systemische Therapien hilfreich sind, können sie unerwünschte Wirkungen verursachen.
In der Regel tritt innerhalb weniger Monate eine Remission ein, manchmal ist jedoch eine Behandlung über mehrere Jahre erforderlich.
Behandlung von bullösem Pemphigoid
Topische oder orale Kortikosteroide
Entzündungshemmende Medikamente
Immunsuppressive Medikamente oder Biologika
Hochpotente topische Kortikosteroide (z. B. Clobetasol 0,05% Creme) sollten für lokalisierte Erkrankungen verwendet werden und können die erforderliche Dosis von systemischen Medikamenten reduzieren.
Bei generalisierter Erkrankung benötigen die Patienten häufig eine systemische Behandlung mit Prednison 60–80 mg oral 1-mal täglich, das nach mehreren Wochen auf eine Erhaltungsdosis von ≤ 10–20 mg täglich ausgeschlichen werden kann. Die meisten Patienten erreichen eine Remission nach 2 bis 10 Monaten, aber die Behandlung muss möglicherweise mehrere Jahre fortgesetzt werden, bevor der Krankheitsprozess ausreichend ist, um das Absetzen zu ermöglichen. Falls eine Langzeittherapie erforderlich ist, wird keine Erhöhung der Prednison-Dosis benötigt, wenn alle paar Wochen eine neue Blase entsteht.
Bullöses Pemphigoid reagiert gelegentlich auf die entzündungshemmende Wirkung bestimmter Medikamente, wie beispielsweise die Kombination von Tetracyclin oder Minocyclin und Nikotinamid. Andere Behandlungsmöglichkeiten umfassen eine Monotherapie mit Dapson, Sulfapyridin oder Erythromycin. I.v. Immunglobulin wurde auch gelegentlich genutzt.
Für Patienten mit generalisierten und hartnäckigen Erkrankungen und manchmal, um die Kortikosteroid-Dosis bei chronischen Erkrankungen zu verringern, können Immunsuppressiva wie Methotrexat, Azathioprin, Cyclophosphamid, Mycophenolatmofetil und Ciclosporin verwendet werden. Unter den Biologika können Rituximab und Omalizumab verwendet werden.
Wichtige Punkte
Das bullöse Pemphigoid betrifft in der Regel Patienten im Alter von > 60 Jahren und ist autoimmun und idiopathisch.
Ein Pruritus kann mehrere Jahre vor der Entwicklung eines Hautausschlages auftreten. Eine Beteiligung der Schleimhaut ist selten.
Es sollten eine Hautbiopsie für die Histologie und für Immunfluoreszenztests sowie eine Messung der zirkulierenden Autoantikörper durchgeführt werden.
Die Behandlung der Patienten sollte, wenn möglich, mit hochpotenten topischen Kortikosteroiden erfolgen, um den Einsatz von systemischen Kortikosteroiden zu verhindern oder zu minimieren.
Eine entzündungshemmende, immunsuppressive und biologische Therapie kann angewendet werden, um die Kortikosteroiddosis zu senken.
Die Symptome nehmen in der Regel innerhalb weniger Monate ab, aber manchmal ist eine Behandlung über mehrere Jahre erforderlich.