




Amyloidose ist eine seltene Erkrankung, bei der abnorm gefaltete Proteine sich zu sogenannten Amyloidfibrillen verketten, die sich in verschiedenen Geweben und Organen ansammeln und mitunter zu Organfehlfunktionen, Organversagen und Tod führen.
Die Symptome und die Schwere der Amyloidose hängt davon ab, welche wichtigen Organe betroffen sind.
Die Diagnose wird durch Entnahme von Gewebeproben (Biopsie) für eine mikroskopische Untersuchung gestellt.
Es gibt viele Formen von Amyloidose und es sind weitere Tests erforderlich, um die Art und ihre Ursache herauszufinden.
Die Behandlung hängt von der Art der Amyloidose ab.
Ursachen von Amyloidose
Proteine sind lange Molekülketten, die sich in einer bestimmten Form zusammenfalten. Die genaue Form ist für die Funktion der einzelnen Proteine entscheidend. Alle Formen von Amyloidosen enthalten ein Protein, das sich abnorm faltet. Die abnorm gefalteten Proteine verklumpen sich und häufen sich in verschiedenen Geweben des Körpers als amyloide Ablagerungen an. Diese Ablagerungen bestehen aus Amyloidfibrillen, die anormale Proteinfasern enthalten, die vom Körper schwer aufzuspalten und abzubauen sind. Diese Ablagerungen beeinträchtigen die normale Funktion des Organs, in dem sie zu finden sind. All diese Proteine werden vom Körper selbst und nicht durch die Ernährung einer Person gebildet. Einige Amyloid-Proteine sind mutierte Versionen normaler Proteine. Andere sind normale Proteine, die lediglich dazu neigen, sich abnorm zu falten. Einige der Proteine werden bei verschiedenen anderen Erkrankungen produziert (z. B. Tuberkulose oder rheumatoider Arthritis ).
Formen von Amyloidose
Amyloidablagerungen können sein:
Systemisch: im ganzen Körper vorkommend
Lokalisiert: nur ein Organ oder Gewebe betreffend
Der Schweregrad der Krankheit hängt davon ab, welche Organe von den Amyloidablagerungen betroffen sind.
Systemische Amyloidose
Eine systemische Amyloidose kann in vier Hauptgruppen unterteilt werden:
Die (primäre) AL-Amyloidose (Leichtkettenamyloidose) entsteht durch Abnormitäten in Plasmazellen (Immunzellen, die Antikörper herstellen), die bewirken, dass sie übermäßige Mengen abnormer Antikörperproteine bilden, die als leichte Ketten bezeichnet werden. Einige Menschen mit AL-Amyloidose haben auch Krebs der Plasmazellen (multiples Myelom). Nur etwa 10 bis 20 Prozent der Menschen mit multiplem Myelom entwickeln eine AL-Amyloidose. Bei der AL-Amyloidose wird das Amyloid hauptsächlich in Haut, Herz, Nieren, Nerven, Zunge, Darm, Leber, Milz und Blutgefäßen eingelagert.
Die Sekundär-Amyloidose kann eine Reaktion auf verschiedene Krankheiten sein, die anhaltende Infektionen oder Entzündungen (wie Tuberkulose, rheumatoide Arthritis und familiäres Mittelmeerfieber) und bestimmte Krebsarten auslösen. Die AA-Amyloidose führt sehr häufig zu Erkrankungen der Nieren, es können aber auch andere Organe betroffen sein.
Die AF- (familiäre) Amyloidose bezeichnet eine Reihe von seltenen Erbkrankheiten, die Symptome im Erwachsenenalter bewirken. Der Amyloid-produzierende Defekt entsteht aufgrund vererbter Mutationen bestimmter Proteine im Blut. Diese mutierten Proteine bilden Amyloidfibrillen aus, die typischerweise auf die Nieren, Nerven oder das Herz abzielen. Mutiertes Transthyretin, ein Protein, das von der Leber gebildet wird, ist die häufigste Ursache für familiäre Amyloidose.
ATTRwt-(Wildtyp-Transthyretin)-Amyloidose (früher senile systemische Amyloidose genannt) betrifft für gewöhnlich das Herz. Es ist tritt viel häufiger bei Männern als bei Frauen auf. ATTRwt-Amyloidose wird durch eine abnorme Faltung von normalem (Wildtyp, nicht mutiertem) Transthyretin-Protein ausgelöst. Es ist nicht bekannt, warum sich Amyloid im Herzen ablagert.
Lokalisierte Amyloidose
Die lokalisierte Amyloidose tritt auf, wenn sich Amyloid in bestimmten Organen oder Geweben ablagert. Amyloidablagerungen finden sich z. B. im Gehirn von Patienten mit Alzheimerkrankheit. Man geht davon aus, dass sie an der Entstehung der Alzheimerkrankheit beteiligt sind. Lokalisierte Amyloidablagerungen sind auch in der Haut, dem Darm, den Atemwegen oder Blase zu finden.
Symptome einer Amyloidose
Der Aufbau großer Mengen von Amyloidablagerungen kann die normale Funktion vieler Organe beeinträchtigen. Manche Menschen haben kaum Symptome. Andere wiederum entwickeln eine schwere, lebensbedrohliche Erkrankung. Häufige Anzeichen der Amyloidose sind Müdigkeit und Gewichtsverlust. Andere Symptome einer Amyloidose hängen davon ab, wo sich die Amyloidablagerungen aufbauen.
Ist das Herz betroffen, können Betroffene Herzrhythmusstörungen oder Herzinsuffizienz haben, die Kurzatmigkeit, Schwäche oder Ohnmacht hervorrufen.
Sind die Nerven betroffen, können Betroffene Kribbeln oder Taubheit in den Fingern oder Zehen verspüren oder Schwindel, wenn sie aufstehen. Sind die Nieren betroffen, können Betroffene an Schwellungen (Ödeme) an den Füßen und Beinen und manchmal im Bauch leiden.

© Springer Science+Business Media
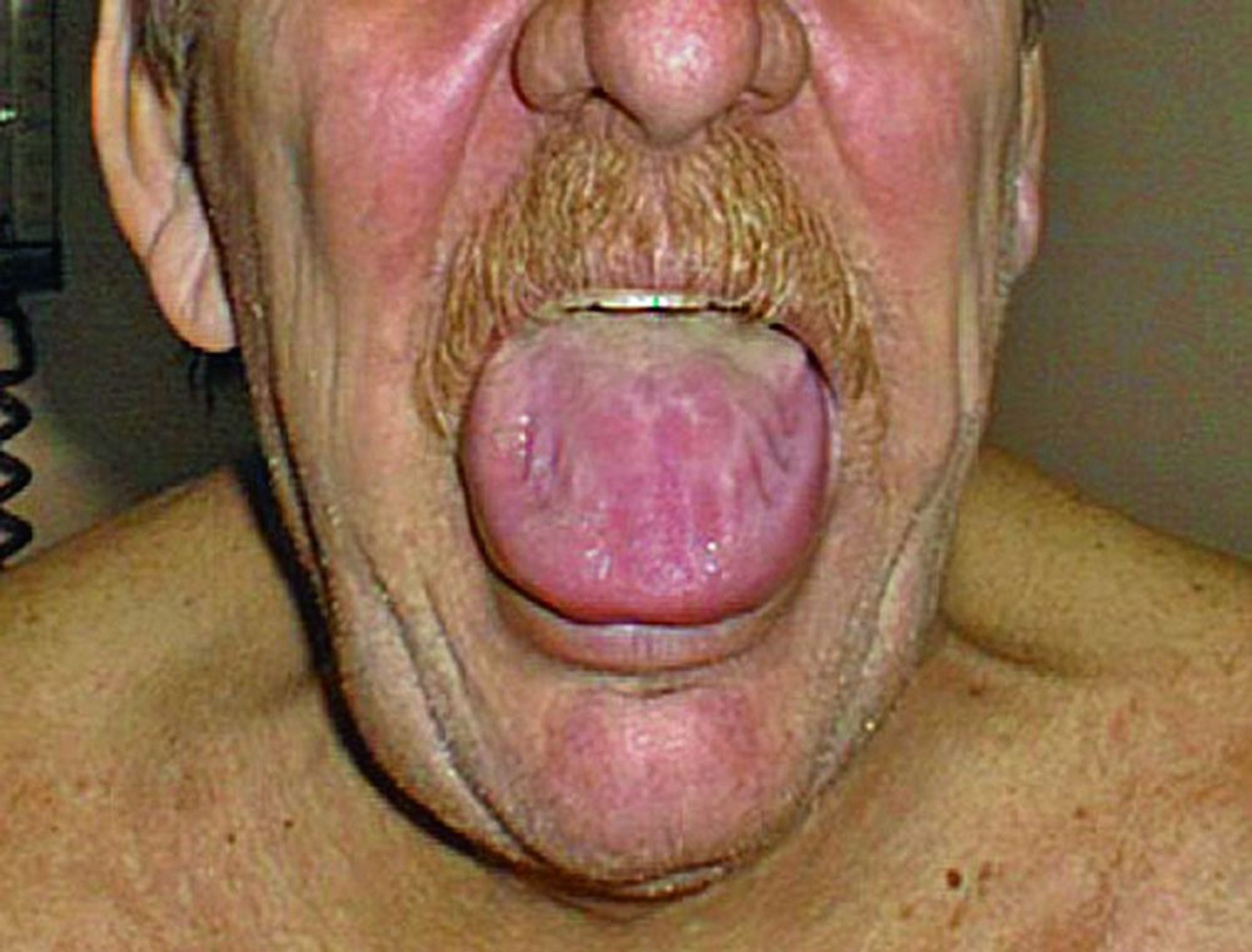
Ist die Haut betroffen, sind blaue Flecken häufig, die mitunter auch um die Augen herum auftreten können. Die Zunge ist manchmal vergrößert (Makroglossie).
Diagnose einer Amyloidose
Biopsie
Manchmal ist eine Amyloidose für die Ärzte schwer zu erkennen, da sie so viele verschiedene Probleme verursacht, die ebenso durch ganz andere Krankheiten entstehen können. Eine Amyloidose wird dann vermutet, wenn Patienten Symptome aufweisen, die mehrere Organe des Körpers betreffen, oder wenn sie an unerklärlicher Herz-, Nieren- oder Leberinsuffizienz leiden. Makroglossie ist kein häufig auftretendes Symptom. Wenn es jedoch auftritt, spricht es für eine Amyloidose. Wenn mehrere Familienmitglieder an Herz- oder Nervenproblemen leiden, kann dies auf eine AF-Amyloidose hinweisen. Unerklärliche Herzsymptome bei älteren Männern deuten auf eine ATTRwt-Amyloidose hin.
Zur Diagnose wird im Allgemeinen etwas Bauchfett mithilfe einer Nadel, die in der Nähe des Bauchnabels in den Bauch gestochen wird, entnommen und untersucht (Fettpolsterbiopsie). Alternativ können Ärzte eine Gewebeprobe von dem Körperteil entnehmen, der von der Amyloidose betroffen ist, etwa dem Herzen, der Nieren oder Leber, und diese mikroskopisch mithilfe bestimmter Farbstoffe untersuchen.
Nachdem der Arzt festgestellt hat, dass der Patient an Amyloidose leidet, führt er Tests zur Bestimmung der genauen Amyloidoseform und zur Ermittlung sämtlicher Krankheiten durch, die Ursache der Amyloidose sein könnten. Zudem werden Tests gemacht, um zu untersuchen, welche Organe betroffen sind.
Behandlung einer Amyloidose
Bei AL-Amyloidose: Chemotherapie
Bei AA-Amyloidose: Behandlung der zugrundeliegenden Ursache
Bei einer Amyloidose, die durch Transthyretin-Proteinablagerungen verursacht wurde: Medikamente zur Stabilisierung von Transthyretin
Manchmal: Organtransplantation
Die Behandlung zur Verringerung oder Kontrolle der Symptome und Komplikationen einer Amyloidose kann die Lebensqualität von Menschen mit jeder Form von Amyloidose verbessern. Bestimmte Behandlungen, die eine Bildung von Amyloid verlangsamen oder stoppen, können bei bestimmten Formen von Amyloidose helfen.
Bei Menschen mit AL-Amyloidose kann eine Chemotherapie mit Melphalan, Bortezomib oder Lenalidomid, manchmal zusammen mit einer peripheren Stammzellentransplantation, die Krankheit im Knochenmark stoppen und weiteren Ablagerungen von Amyloid vorbeugen. Eine Strahlentherapie kann bei Menschen angewendet werden, deren AL-Amyloidose in nur einem Bereich vorliegt (lokalisierte Erkrankung).
Bei der Sekundär-Amyloidose kann die Behandlung der zugrundeliegenden Ursache die Amyloidablagerungen reduzieren. Bei einer AA-Amyloidose, die durch familiäres Mittelmeerfieber verursacht wurde, wirkt Colchicin sehr gut.
Bei einer Amyloidose, die durch Transthyretin-Proteinablagerungen verursacht wurde, können Medikamente wie Diflunisal und Tafamidis das mutierte Transthyretin-Protein stabilisieren (es also von der Bildung von Amyloidfibrillen abhalten) und damit ein Fortschreiten der vererbten oder der Wildtyp–Transthyretin-Krankheitsform verlangsamen. Gentherapien, die die Transthyretin-Produktion (wie Patisiran, Inotersen und Vutrisiran) reduzieren, können die Auswirkungen der vererbten Krankheitsform auf das Nervensystem verringern.
Organtransplantationen (z. B. von Niere oder Herz) haben bei einigen Menschen mit Organversagen aufgrund von Amyloidose das Leben verlängert.
Bei der familiären Transthyretin-Amyloidose kann eine Lebertransplantation vorgenommen werden. Eine Lebertransplantation kann das Fortschreiten der Krankheit verlangsamen, weil in der Leber das mutierte Protein gebildet wird. Interessanterweise werden aufgrund des Mangels an Spenderorganen die Leberorgane, die Personen mit familiärer Transthyretin-Amyloidose entnommen wurden, manchmal in Menschen mit tödlichen Lebererkrankungen wie Zirrhose oder Leberkrebs transplantiert. Eine derartige „Dominotransplantation“ ist möglich, weil eine Leber von einer Person mit familiärer Transthyretin-Amyloidose eine ansonsten normal funktionierende Leber ist. Auch wenn Menschen, die von jemandem mit familiärer Transthyretin-Amyloidose eine Leber gespendet bekamen, selbst irgendwann eine Amyloidose entwickeln könnten, kann das transplantierte Organ zumindest kurzfristig ihr Leben retten.
Prognose bei Amyloidose
Die Prognose hängt von der Art der Amyloidose und dem betroffenen Organsystem ab. Eine Beteiligung des Herzens ist die gefährlichste Form und kann eine düstere Prognose mit sich bringen.