


Congenital anomalies of the urethra in boys usually involve anatomic abnormalities of the penis and vice versa. In girls, urethral anomalies may exist without other external genital abnormalities. Surgical repair is needed when function is impaired or cosmetic correction is desired.
Penile Anomalies
Chordee
This anomaly is ventral, lateral, and/or rotational curvature of the penis, which is most apparent with erection and is caused by fibrous tissue along the usual course of the corpus spongiosum, or by a size difference between the two corpora cavernosa. Chordee may be associated with hypospadias. Severe deformity may require surgical correction.
Epispadias
The urethra opens on the dorsum of the glans or penile shaft, or at the penopubic junction. In girls, the urethra opens between the clitoris and labia or in the abdomen. Epispadias can be partial (in 15%) or complete; the most severe form occurs with bladder exstrophy. Symptoms and signs of epispadias are incontinence, urinary reflux, and urinary tract infections (UTIs).
Treatment of epispadias is surgical. In partial epispadias, prognosis for continence with treatment is good. In complete epispadias, surgical reconstruction of the penis alone may lead to persistent incontinence; bladder outlet reconstruction is required to achieve complete urinary control.

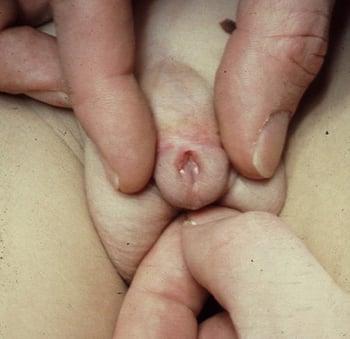
Hypospadias
This anomaly is caused by failure of tubularization and fusion of the urethral groove. It almost always occurs in boys, in whom the urethra opens onto the underside of the penile shaft, at the penoscrotal junction, between the scrotal folds, or in the perineum. The foreskin fails to become circumferential and appears as a dorsal hood. Hypospadias is frequently associated with chordee.
Prognosis for functional and cosmetic correction is good. Outpatient surgery at about 6 months of age involves construction of a neourethra using penile shaft skin or foreskin and repair of the chordee.
Hypospadias is extremely rare in girls; the urethra opens into the vaginal introitus.
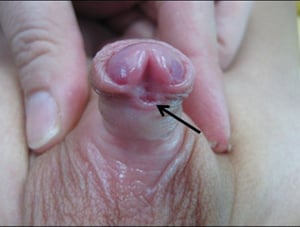
Image courtesy of Drs. Ronald Rabinowitz and Jimena Cubillos.
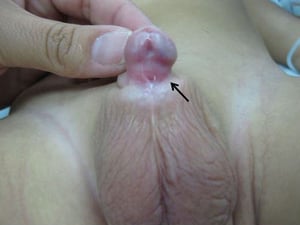
Image courtesy of Drs. Ronald Rabinowitz and Jimena Cubillos.
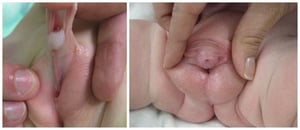

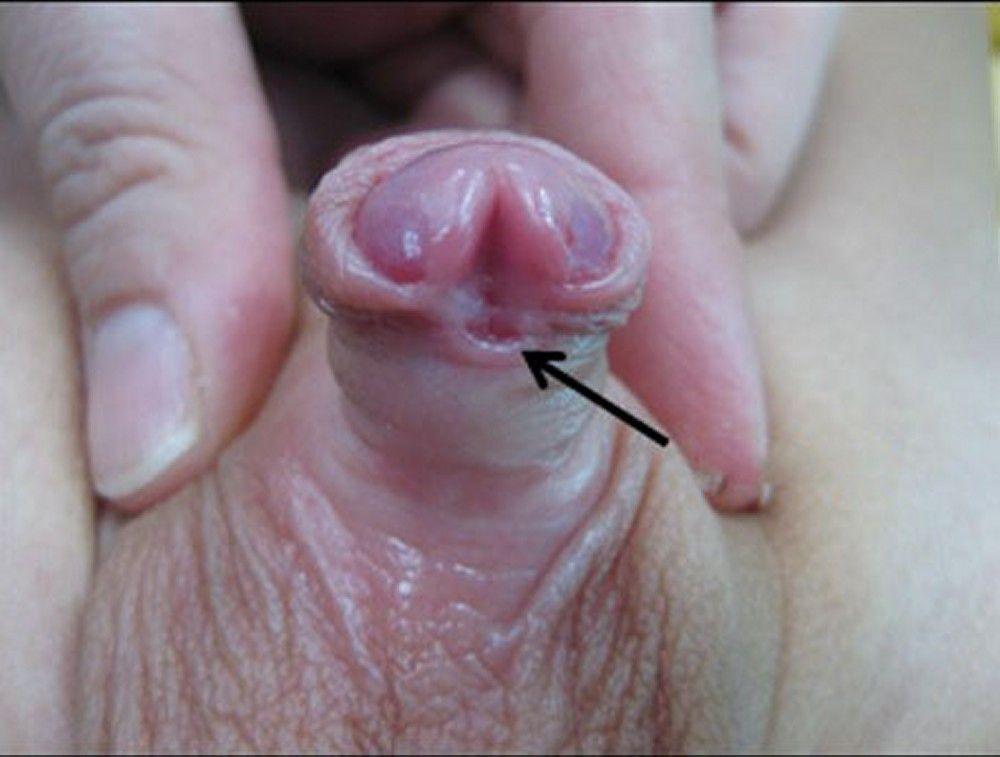
Image courtesy of Drs. Ronald Rabinowitz and Jimena Cubillos.

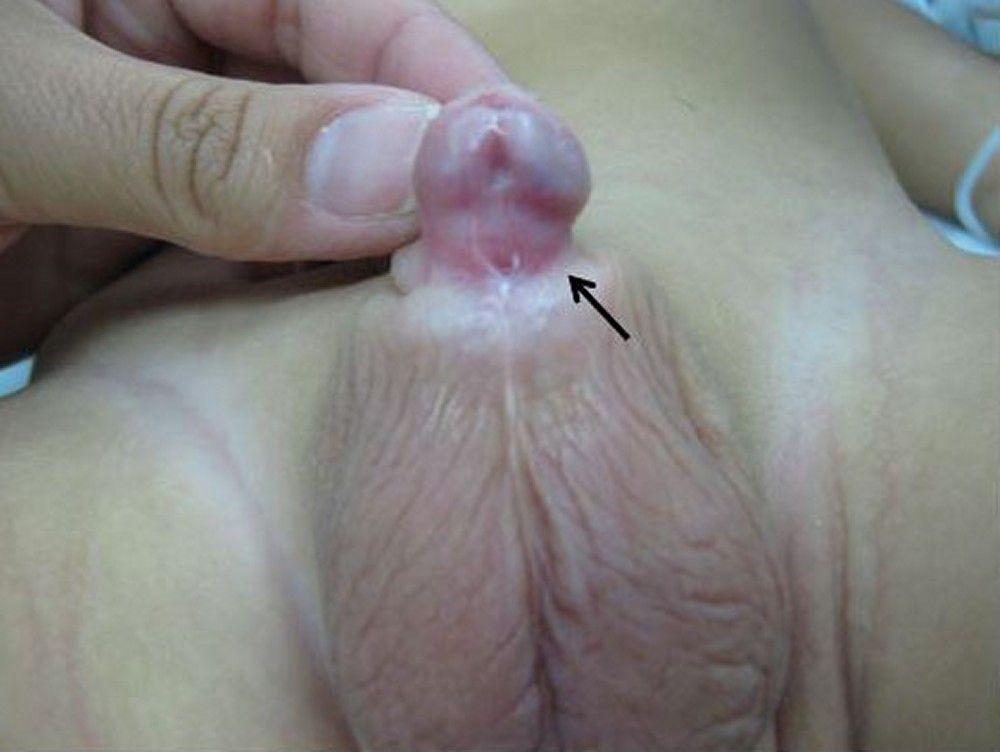
Image courtesy of Drs. Ronald Rabinowitz and Jimena Cubillos.

Phimosis and paraphimosis
Phimosis, the most common penile abnormality, is constriction of the foreskin with inability to retract over the glans; it may be congenital or acquired.
Paraphimosis is inability of the retracted constricting foreskin to be reduced distally over the glans.
Phimosis may respond to topical corticosteroids and gentle stretching; some boys require circumcision.
Paraphimosis should be reduced urgently because the constricting foreskin functions as a tourniquet, causing edema and pain. Firm circumferential compression of the edematous foreskin with the fingers may reduce edema sufficiently to allow the foreskin to be restored to its normal position by pushing the glans back through the tight foreskin using both thumbs. If this technique is ineffective, a dorsal slit done using a local anesthetic relieves the condition temporarily. When edema has resolved, the phimosis may be treated with circumcision or topical corticosteroids.
Other penile anomalies
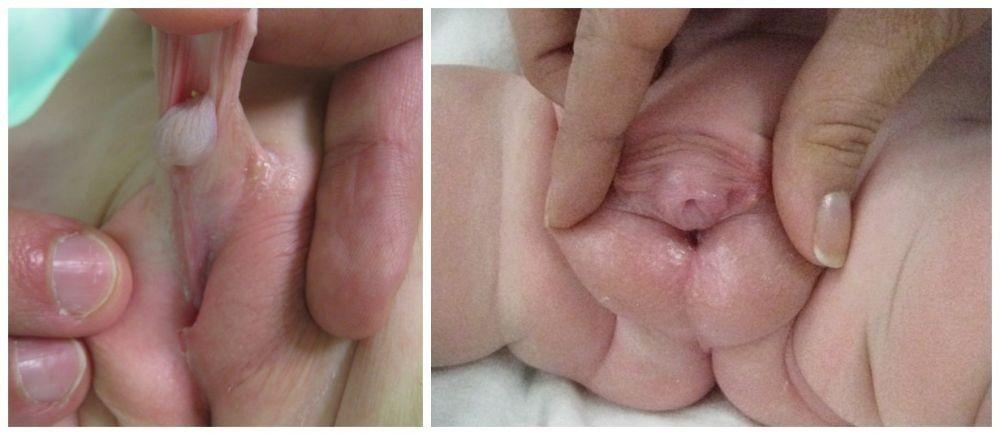
A very tight frenulum may prevent complete retraction of the foreskin or cause pain or bleeding with foreskin retraction or erection. Frenulectomy may be sufficient to resolve symptoms if patients do not want circumcision.
Less common anomalies include penile agenesis, duplication, and lymphedema. Many anomalies also involve urethral abnormality, or other anomalies, such as exstrophy. Treatment of most anomalies is surgical.
Urethral Anomalies
Urethral meatal stenosis
Most commonly acquired after circumcision in newborn boys, urethral meatal stenosis is occasionally congenital and associated with hypospadias. Meatotomy is needed for a significantly deflected stream or for a pinpoint stream.
Urethral stricture
Urethral stricture causes obstruction along some part of the length of the urethra. It almost always occurs in boys, is usually acquired, and typically results from a crush injury after straddle trauma. Urethral strictures may also be iatrogenic and occur after surgery, most commonly after hypospadias repair, or after traumatic catheterization. Congenital urethral stricture may manifest similarly to urethral valves and may be diagnosed by prenatal ultrasonography, or postnatally by symptoms and signs of outlet obstruction or patent urachus and is confirmed by retrograde urethrography. Initial management is often with endoscopic urethrotomy, although open urethroplasty may be necessary.
Urethral duplications and triplications are urethral anomalies. The patent urethra is the most ventral channel. Voiding cystourethrography (VCUG) should be done to determine patency and detect connection between the channels and the bladder. Surgical intervention is almost always necessary.
Urethral valves
In boys, folds in the posterior urethra may act as valves impairing urine flow. Urologic sequelae of urethral valves include urinary hesitancy, decreased urinary stream, urinary tract infection, overflow incontinence, myogenic bladder malfunction, vesicoureteral reflux, upper urinary tract damage, and renal insufficiency. The valves occasionally occur with a patent urachus. Because fetal urine excretion contributes to the amniotic fluid, severe urethral obstruction can cause decreased amniotic fluid (oligohydramnios), which can cause lung hypoplasia and consequent pulmonary hypertension, pulmonary hypoplasia, and/or respiratory failure. Pulmonary hypertension can then cause systemic hypertension. Severe cases may result in perinatal demise.
Diagnosis is often made by findings on routine prenatal ultrasonography, including severe bilateral hydroureteronephrosis or oligohydramnios. Cases suspected postnatally (often because of history of an abnormal urine stream) are confirmed by immediate voiding cystourethrography.
Surgery (usually via endoscopy) is done at time of diagnosis to prevent progressive renal deterioration. If endoscopic valve ablation is not feasible, a temporary vesicostomy is indicated.
A much less common anomaly, diverticulum of the anterior urethra, may act as a valve (anterior urethral valve). This anomaly is frequently treated endoscopically and occasionally by open reconstruction.

Urethral prolapse
Urethral prolapse is protrusion of a small portion of the distal urethra outside the meatus. Prolapse occurs mainly in girls, most commonly in Black girls. It may appear in infancy or later in childhood and usually manifests as an interlabial mass. When urethral prolapse occurs, the opening of the urethra resembles a donut with the meatus located in the center. It may become irritated and cause pain and/or bleeding. Treatment is usually conservative with a topical estrogen cream. Surgical intervention is rarely necessary.






