


Congenital heart disease is the most common congenital anomaly, occurring in almost 1% of live births (1). Among birth defects, congenital heart disease is the leading cause of infant mortality.
The most common congenital heart diseases diagnosed in infancy are muscular and perimembranous ventricular septal defects followed by secundum atrial septal defects, with a total prevalence of 48.4 in 10,000 live births (2, 3, 4). The most common cyanotic congenital heart disease is tetralogy of Fallot, which is twice as prevalent as transposition of the great arteries (4.7 vs. 2.3/10 000 births). Overall, bicuspid aortic valves are the most common congenital defects with a prevalence reported to be as high as 0.5% to 2.0%.
General references
1. Reller MD, Strickland MJ, Riehle-Colarusso T, et al: Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr 153(6):807–813, 2008.
2. Freeze SL, Landis BJ, Ware SM, Helm BM: Bicuspid aortic valve: a review with recommendations for genetic counseling. J Genet Couns 25(6):1171–1178, 2016.
3. van der Linde D, Konings EEM, Slager MA, et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247, 2011. doi: 10.1016/j.jacc.2011.08.025
4. Daubeney PEF, Rigby ML, Niwa K, Gatzoulis MA (eds): Pediatric Heart Disease: A Practical Guide. Wiley-Blackwell 2012 .
Etiology of Congenital Heart Disease
Environmental and genetic factors contribute to the development of congenital heart disease.
Common environmental factorsDown syndrome, that may include cardiac defects. It is unclear whether maternal age is an independent risk factor for congenital heart disease. Paternal age may also be a risk factor (1).
Certain numerical chromosomal abnormalities (aneuploidies), such as trisomy 21 (Down syndrome), trisomy 18, trisomy 13, and monosomy X (Turner syndrome), are strongly associated with congenital heart disease. However, these abnormalities account for only 5 to 6% of patients with congenital heart disease.
Many other cases involve subchromosomal deletions (microdeletions), subchromosomal duplications, or single-gene mutations. Often, these mutations cause congenital syndromes affecting multiple organs in addition to the heart. Examples include DiGeorge syndrome (microdeletion in 22q11.2) and Williams (sometimes known as Williams-Beuren) syndrome (microdeletion in 7p11.23). Single-gene defects that cause syndromes associated with congenital heart disease include mutations in fibrillin-1 (Marfan syndrome), TXB5 (Holt-Oram syndrome), and PTPN11 (Noonan syndrome). Single-gene defects can also cause isolated (ie, nonsyndromic) congenital heart defects.
No identifiable genetic etiology is detected in about 72% of patients with congenital heart disease (2, 3, 4).
The recurrence risk of congenital heart disease in a family depends on the cause. Risk is negligible in de novo mutations, 2 to 5% in nonsyndromic multifactorial congenital heart disease, and 50% when an autosomal dominant mutation is the cause. The identification of a bicuspid aortic valve in an individual merits family screening in view of the reported familial prevalence of 9% (5). It is important to identify genetic factors because more patients with congenital heart disease are surviving into adulthood and potentially starting families.
Etiology references
1. Materna-Kiryluk A, Wiśniewska K, Badura-Stronka M, et al: Parental age as a risk factor for isolated congenital malformations in a Polish population. Paediatr Perinat Epidemiol 23(1):29-40, 2009. doi: 10.1111/j.1365-3016. 2008.00979.x
2. Russell MW, Chung WK, Kaltman JR, Miller TA: Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes. J Am Heart Assoc 7(6):e006906, 2018. doi:10.1161/JAHA.117.006906
3. van der Linde D, Konings EEM, Slager MA, et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247, 2011. doi: 10.1016/j.jacc.2011.08.025
4. Pierpont ME, Brueckner M, Chung WK, et al: Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association [published correction appears in Circulation 2018 Nov 20;138(21):e713]. Circulation 138(21):e653–e711, 2018. doi:10.1161/CIR.0000000000000606
5. Freeze SL, Landis BJ, Ware SM, Helm BM: Bicuspid aortic valve: a review with recommendations for genetic counseling. J Genet Couns 25(6):1171–1178, 2016.
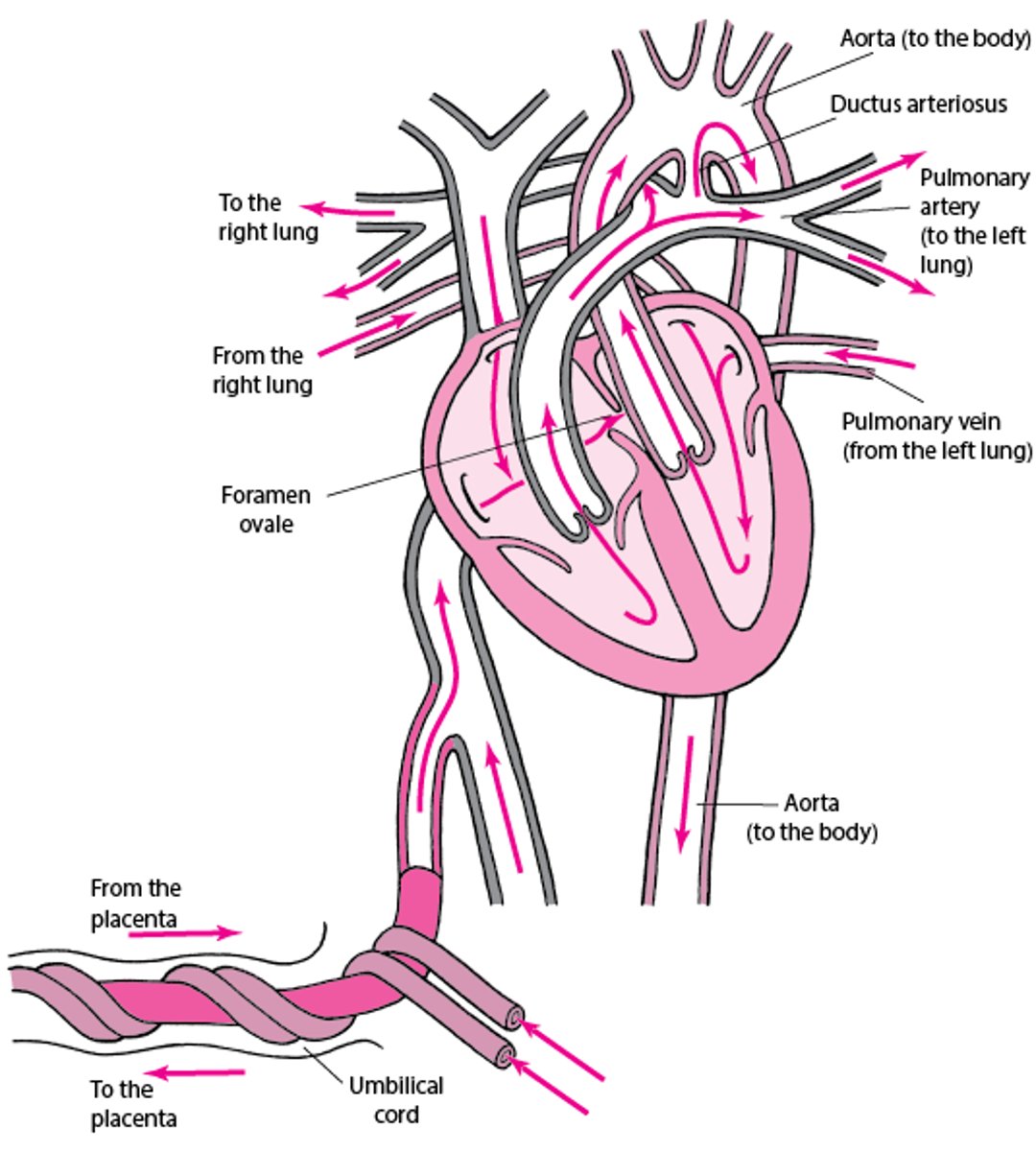
Normal Fetal Circulation
Fetal circulation is marked by
Right-to-left shunting of blood around the unventilated lungs through a patent ductus arteriosus (connecting the pulmonary artery to the aorta) and foramen ovale (connecting the right and left atria)
Shunting is encouraged by high pulmonary arteriolar resistance and relatively low resistance to blood flow in the systemic (including placental) circulation. About 90 to 95% of the right heart output bypasses the lungs and goes directly to the systemic circulation. The fetal ductus arteriosus is kept open by low fetal systemic PaO2 (about 25 mm Hg) along with locally produced prostaglandins. The foramen ovale is kept open by differences in atrial pressures: left atrial pressure is relatively low because little blood is returned from the lungs, but right atrial pressure is relatively high because large volumes of blood return from the placenta.
Normal circulation in a fetus
In the fetus, blood entering the right side of the heart has already been oxygenated via the placenta. Because the lungs are not ventilated, only a small amount of blood needs to go through the pulmonary artery. Most of the blood from the right side of the heart bypasses the lungs through the
Normally, these two structures close shortly after birth. |

Perinatal changes
Profound changes to this system occur after the first few breaths, resulting in
Increased pulmonary blood flow
Functional closure of the foramen ovale
Pulmonary arteriolar resistance drops acutely as a result of vasodilation caused by lung expansion, increased PaO2, and reduced PaCO2. The elastic forces of the ribs and chest wall decrease pulmonary interstitial pressure, further enhancing blood flow through pulmonary capillaries. Increased venous return from the lungs raises left atrial pressure, thus reducing the pressure differential between left and right atria; this effect contributes to the functional closure of the foramen ovale.
As pulmonary blood flow is established, venous return from the lungs increases, raising left atrial pressure. Air breathing increases the PaO2, which constricts the umbilical arteries. Placental blood flow is reduced or stops, reducing blood return to the right atrium. Thus, right atrial pressure decreases while left atrial pressure increases; as a result, the 2 fetal components of the interatrial septum (septum primum and septum secundum) are pushed together, stopping flow through the foramen ovale. In most people, the 2 septa eventually fuse and the foramen ovale ceases to exist. However, in 25% of adults, the foramen ovale may remain patent with minimal or no residual shunting (1).
Soon after birth, systemic resistance becomes higher than pulmonary resistance, a reversal from the fetal state. Therefore, the direction of blood flow through the patent ductus arteriosus reverses, creating left-to-right shunting of blood (called transitional circulation). This state lasts from moments after birth (when the pulmonary blood flow increases and functional closure of the foramen ovale occurs) until about 24 to 72 hours of age, when the ductus arteriosus constricts. Blood entering the ductus and its vasa vasorum from the aorta has a high PO2, which, along with alterations in prostaglandin metabolism, leads to constriction and closure of the ductus arteriosus. Once the ductus arteriosus closes, an adult-type circulation exists. The 2 ventricles now pump in series, and there are no major shunts between the pulmonary and systemic circulations.
During the days immediately after birth, a stressed neonate may revert to a fetal-type circulation. Asphyxia with hypoxia and hypercarbia causes the pulmonary arterioles to constrict and the ductus arteriosus to dilate, reversing the processes described previously and resulting in right-to-left shunting through the now-patent ductus arteriosus, the reopened foramen ovale, or both. Consequently, the neonate becomes severely hypoxemic, a condition called persistent pulmonary hypertension or persistent fetal circulation (although there is no umbilical circulation). The goal of treatment is to reverse the conditions that caused pulmonary vasoconstriction.
Normal fetal circulation reference
1. Koutroulou I, Tsivgoulis G, Tsalikakis D, et al: Epidemiology of Patent Foramen Ovale in General Population and in Stroke Patients: A Narrative Review. Front Neurol 11:281, 2020. Published 2020 Apr 28. doi:10.3389/fneur.2020.00281
Pathophysiology of Congenital Heart Anomalies
Congenital heart anomalies are classified (see table Classification of Congenital Heart Anomalies) as
Cyanotic
Acyanotic (left-to-right shunts or obstructive lesions)
The physiologic consequences of congenital heart anomalies vary greatly, ranging from a heart murmur or discrepancy in pulses in an asymptomatic child to severe cyanosis, heart failure, or circulatory collapse.
Classification of Congenital Heart Anomalies*
Classification | Examples |
|---|---|
Cyanotic | |
— | |
Acyanotic | |
Left-to-right shunt | |
Obstructive | Hypoplastic left heart syndrome (often also manifests with cyanosis, which may be mild) |
* In approximate decreasing order of frequency. | |
Cyanotic heart anomalies
Varying amounts of deoxygenated venous blood are shunted to the left heart (right-to-left shunt), reducing systemic arterial oxygen saturation.
If there is > 5 g/dL (> 50 g/L) of deoxygenated hemoglobin, cyanosis results. Complications of persistent cyanosis include polycythemia, clubbing, thromboembolism (including stroke), bleeding disorders, brain abscess, and hyperuricemia. Hypercyanotic spells can occur in infants with unrepaired tetralogy of Fallot or other complex congenital defects with dynamic subpulmonic stenosis and a ventricular defect.
Depending on the anomaly, pulmonary blood flow may be reduced, normal, or increased (often resulting in heart failure in addition to cyanosis), resulting in cyanosis of variable severity. Heart murmurs are variably audible and are not specific.
Left-to-right shunts
Oxygenated blood from the left heart (left atrium or left ventricle) or the aorta shunts to the right heart (right atrium or right ventricle) or the pulmonary artery through an opening or communication between the 2 sides.
Immediately after birth, pulmonary vascular resistance is high and flow through this communication may be minimal or bidirectional. Within the first 24 to 48 hours of life, however, the pulmonary vascular resistance progressively falls, at which point blood will increasingly flow from left to right. The additional blood flow to the right side increases pulmonary blood flow and pulmonary artery pressure to a varying degree. The greater the increase, the more severe the symptoms; a small left-to-right shunt typically does not cause symptoms or signs.
High-pressure shunts (those at the ventricular or great artery level) become apparent several days to a few weeks after birth; low-pressure shunts (atrial septal defects) become apparent considerably later. If untreated, elevated pulmonary blood flow and pulmonary artery pressure may lead to pulmonary vascular disease and eventually Eisenmenger syndrome. Large left-to-right shunts (eg, large ventricular septal defect [VSD], patent ductus arteriosus [PDA]) cause excess pulmonary blood flow and left ventricular volume overload, which may lead to signs of heart failure and during infancy often result in failure to thrive. A large left-to-right shunt also leads to lower lung compliance and higher airway resistance. These factors increase the likelihood of hospitalization in infants with respiratory syncytial virus or other upper or lower respiratory tract infections.
Obstructive lesions
Blood flow is obstructed, causing a pressure gradient across the obstruction.
The resulting pressure overload proximal to the obstruction may cause ventricular hypertrophy and heart failure. The most obvious manifestation is a heart murmur, which results from turbulent flow through the obstructed (stenotic) point. Examples are congenital aortic stenosis, which accounts for 3 to 6% of congenital heart anomalies, and congenital pulmonic stenosis, which accounts for 8 to 12% (1, 2).
Heart failure
Some congenital heart anomalies (eg, bicuspid aortic valve, mild aortic stenosis) do not significantly alter hemodynamics. Other anomalies cause pressure or volume overload, sometimes causing heart failure. Heart failure occurs when cardiac output is insufficient to meet the body’s metabolic needs or when the heart cannot adequately handle venous return, causing pulmonary congestion (in left ventricular failure), edema primarily in dependent tissues and abdominal viscera (in right ventricular failure), or both. Heart failure in infants and children has many causes other than congenital heart anomalies (see table Common Causes of Heart Failure in Children).
Duct-dependent congenital heart disease
The ductus arteriosus is a normal connection between the pulmonary artery and aorta; it is necessary for proper fetal circulation. At birth, the rise in PaO2 and decline in prostaglandin concentration cause closure of the ductus arteriosus, typically beginning within the first 10 to 15 hours of life.
Some congenital cardiac disorders are dependent on the ductus arteriosus remaining open to maintain systemic blood flow (eg, hypoplastic left heart syndrome, critical aortic stenosis, coarctation of the aorta) or pulmonary blood flow (cyanotic lesions such as pulmonary atresia or severe tetralogy of Fallot). Keeping the ductus arteriosus open with exogenous prostaglandin infusion is therefore vital in these disorders prior to definitive repair (usually surgery).
Pathophysiology references
1. Daubeney PEF, Rigby ML, Niwa K, Gatzoulis MA (eds): Pediatric Heart Disease: A Practical Guide. Wiley-Blackwell 2012 .
2. Hoffman JI, Kaplan S: The incidence of congenital heart disease. J Am Coll Cardiol 39(12):1890-1900, 2002. doi:10.1016/s0735-1097(02)01886-7
Symptoms and Signs of Congenital Heart Disease
Manifestations of congenital heart diseases are varied but commonly include
Murmurs
Cyanosis
Heart failure
Diminished or nonpalpable pulses
Other physical examination abnormalities may include circulatory shock, poor perfusion, abnormal 2nd heart sound (S2—single or widely split), systolic click, gallop, or abnormally slow, fast, or irregular rhythm.
Murmurs
Most left-to-right shunts and obstructive lesions cause systolic murmurs. Systolic murmurs and thrills are most prominent at the surface closest to their point of origin, making location diagnostically helpful. Increased flow across the pulmonary or aortic valve causes a midsystolic crescendo-decrescendo (ejection systolic) murmur. Regurgitant flow through an atrioventricular valve or flow across a ventricular septal defect causes a holosystolic (pansystolic) murmur that obscures the first heart sound (S1) as its intensity increases.
Patent ductus arteriosus typically causes a continuous murmur that is uninterrupted by the S2 because blood flows through the ductus during systole and diastole. This murmur is 2-toned, having a more pronounced sound during systole (when driven by higher pressure) than during diastole.
Cyanosis

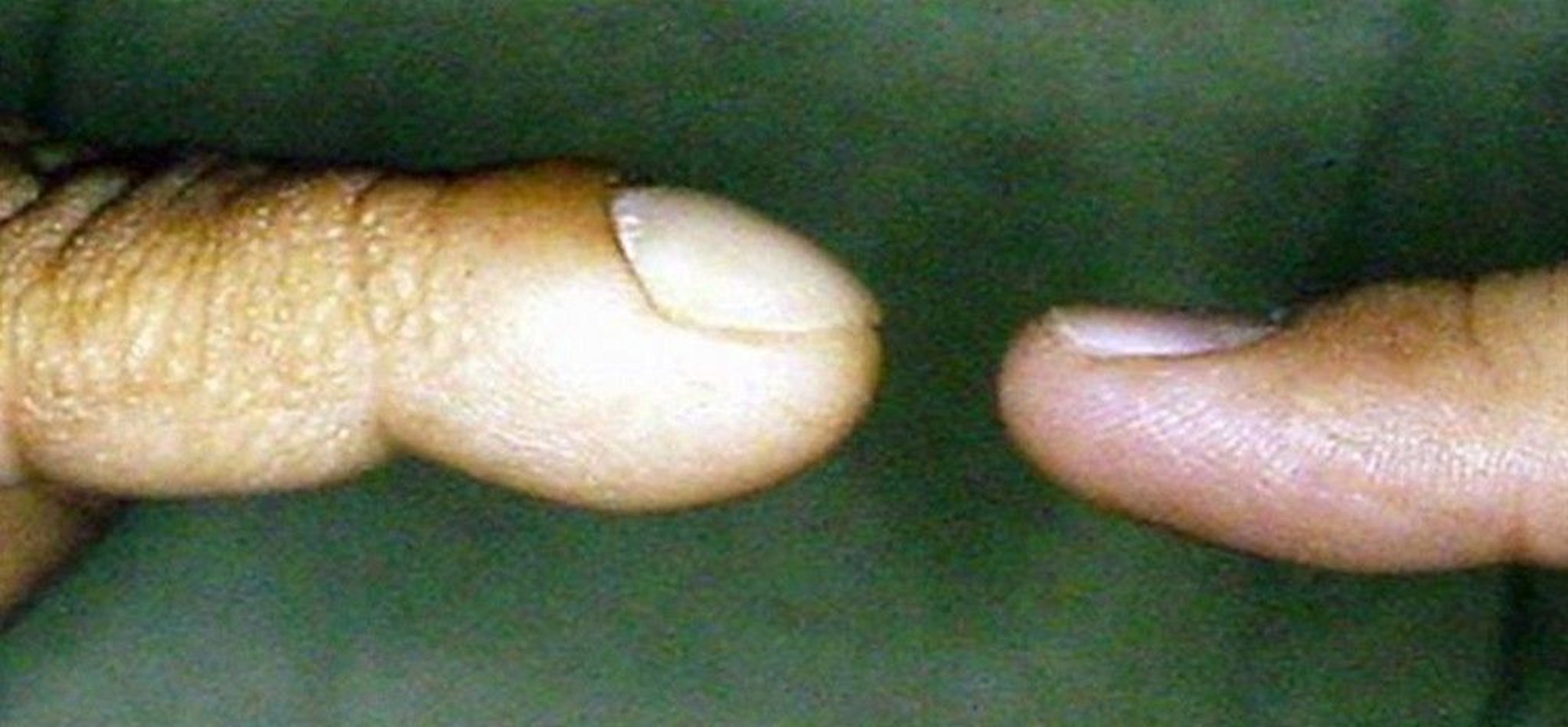
© Springer Science+Business Media
Central cyanosis is characterized by bluish discoloration of the lips and tongue and/or nail beds; it occurs when there is an increase in deoxygenated hemoglobin (at least 5 g/dL [50 g/L]) and implies a low blood oxygen level (usually oxygen saturation < 85%). Perioral cyanosis and acrocyanosis (cyanosis of the hands and feet) without lip or nail bed cyanosis is caused by peripheral vasoconstriction rather than hypoxemia and is a common, normal finding in neonates. Older children with longstanding cyanosis often develop clubbing of the nail beds.
Heart failure
In infants, symptoms or signs of heart failure include
Tachycardia
Tachypnea
Dyspnea during feeding
Diaphoresis, especially during feeding
Restlessness, irritability
Hepatomegaly
Dyspnea during feeding causes inadequate intake and poor growth, which may be worsened by increased metabolic demands in heart failure and frequent respiratory tract infections. In contrast to adults and older children, most infants do not have distended neck veins and dependent edema; however, they occasionally have edema in the periorbital area. Hepatomegaly is a particularly prominent feature of heart failure in infants because of the distensibility of the liver capsule at this age. Findings in older children with heart failure are similar to those in adults.
Other manifestations of heart defects
In neonates, circulatory shock may be the first manifestation of certain anomalies (eg, hypoplastic left heart syndrome, critical aortic stenosis, interrupted aortic arch, coarctation of the aorta). Neonates appear extremely ill with pale or cyanotic mucous membranes, cold extremities, diminished pulses, low blood pressure, and reduced response to stimuli.
Chest pain in children is usually noncardiac. In infants, chest pain may be manifested by unexplained marked irritability, particularly during or after feeding, and can be caused by anomalous origin of the left coronary artery from the pulmonary artery. In older children and adolescents, chest pain due to a cardiac etiology is usually associated with exertion and may be caused by a coronary anomaly, pericarditis, myocarditis, hypertrophic cardiomyopathy, or severe aortic stenosis.
Syncope, typically without warning symptoms and often in association with exertion, may occur with certain anomalies including cardiomyopathy (hypertrophic or dilated), anomalous origin of a coronary artery, or inherited arrhythmia syndromes (eg, long QT syndrome, catecholaminergic polymorphic ventricular tachycardia [CPVT], Brugada syndrome). High school–age athletes are most commonly affected.
Diagnosis of Congenital Heart Disease
Screening by pulse oximetry
Cardiac physical examination
Chest x-ray and ECG
Echocardiography
Sometimes cardiac MRI or CT angiography, cardiac catheterization with angiocardiography
When present, heart murmurs, cyanosis, abnormal pulses, or manifestations of heart failure suggest congenital heart disease. In neonates with these findings, echocardiography is done to confirm the diagnosis of congenital heart disease. If the only abnormality is cyanosis, methemoglobinemia also should be ruled out.
Although echocardiography is typically diagnostic, in select cases, cardiac MRI or CT angiography may clarify important anatomic details. Cardiac catheterization with angiocardiography is occasionally needed to confirm the diagnosis or to assess severity of the anomaly; however, it is generally done only for therapeutic purposes.
Newborn Screening for Congenital Heart Disease
Manifestations of congenital heart disease may be subtle or absent in neonates, and failure or delay in detecting critical congenital heart disease, particularly in the 10 to 15% of neonates who require surgical or inpatient medical treatment in the first hours or days of life, may lead to neonatal mortality or significant morbidity. Thus, universal screening for critical congenital heart disease using pulse oximetry is recommended for all neonates before hospital discharge (1). The screening is done when infants are ≥ 24 hours old and is considered positive if ≥ 1 of the following is present:
Any oxygen saturation measurement is < 90%.
The oxygen saturation measurements in both the right hand and foot are < 95% on 3 separate measurements taken 1 hour apart.
There is > 3% absolute difference between the oxygen saturation in the right hand (preductal) and foot (postductal) on 3 separate, paired measurements taken 1 hour apart.
All neonates with a positive screening result should undergo a comprehensive evaluation for congenital heart disease and other causes of hypoxemia (eg, various respiratory disorders, central nervous system depression, sepsis) typically including a chest x-ray, ECG, echocardiography, and often blood testing. Sensitivity of pulse oximetry screening is slightly > 75%; the congenital heart disease lesions most often missed are left heart obstructive lesions (eg, coarctation of the aorta).
Newborn screening reference
1. Martin GR, Ewer AK, Gaviglio A, et al: Updated Strategies for Pulse Oximetry Screening for Critical Congenital Heart Disease. Pediatrics 146(1):e20191650, 2020. doi:10.1542/peds.2019-1650
Treatment of Congenital Heart Disease
Surgical repair or transcatheter intervention
Treatment of heart failure varies widely depending on the etiology. Definitive therapy typically requires correction of the underlying problem.
After medical stabilization of acute heart failure symptoms or cyanosis, most children require surgical or transcatheter repair; the exceptions are certain ventricular septal defects that are likely to become smaller or close with time or mild valve dysfunction. Transcatheter procedures include
Balloon atrial septostomy for palliation of severely cyanotic neonates with transposition of the great arteries
Balloon dilation of severe aortic valve stenosis or pulmonic valve stenosis
Transcatheter closure of cardiac shunts (most often atrial septal defect and patent ductus arteriosus)
Transcatheter placement of pulmonary valve
Balloon dilation with or without stenting of vascular stenoses, most commonly pulmonary artery stenosis
Heart failure in neonates
Acute, severe heart failure or cyanosis during the first week of life is a medical emergency. Secure vascular access should be established, preferably via an umbilical venous catheter.
When critical congenital heart disease is suspected or confirmed, an IV infusion of prostaglandin E1 should be started at 0.05 to 0.1 mcg/kg/minute. Keeping the ductus open is important because most cardiac lesions manifesting at this age are ductal-dependent for either systemic blood flow (eg, hypoplastic left heart syndrome, critical aortic stenosis, coarctation of the aorta) or pulmonary blood flow (cyanotic lesions such as critical pulmonary stenosis, pulmonary atresia or severe tetralogy of Fallot).
Mechanical ventilation is often necessary in critically ill neonates. Supplemental oxygen should be given judiciously or even withheld because supplemental oxygen can decrease pulmonary vascular resistance, which is harmful to infants with certain defects (eg, hypoplastic left heart syndrome).
Heart failure in older infants and children
1).
23). However, if Wolff-Parkinson-White syndrome
Supplemental oxygen may lessen hypoxemia and alleviate respiratory distress in heart failure; when possible, fractional inspired oxygen (FIO2) should be kept < 40% to minimize the risk of pulmonary epithelial damage. Supplemental oxygen must be used with caution, if at all, in patients with left-to-right shunt lesions or left heart obstructive disease because it may exacerbate pulmonary overcirculation.
In general, a healthy diet, including salt restriction, is recommended, although dietary modifications may be needed depending on the specific disorder and manifestations. Heart failure increases metabolic demands and the associated dyspnea makes feeding more difficult. In infants with critical congenital heart disease, particularly those with left heart obstructive lesions, feedings may be withheld to minimize the risk of necrotizing enterocolitis. In infants with heart failure due to left-to-right-shunt lesions, enhanced caloric content feedings are recommended; these feedings increase calories supplied and do so with less risk of volume overload. Some children require tube feedings to maintain growth. If these measures do not result in weight gain, surgical repair of the anomaly is indicated.
Endocarditis prophylaxis
Guidelines of the American Heart Association for prevention of endocarditis (4) state that antibiotic prophylaxis is required for children with congenital heart disease who have the following:
Unrepaired cyanotic congenital heart disease (including children with palliative shunts and conduits)
Completely repaired congenital heart disease during the first 6 months after surgery if prosthetic material or a device was used
Repaired congenital heart disease with residual defects at or adjacent to the site of a prosthetic patch or prosthetic device
Mechanical or bioprosthetic valve
Previous episode of endocarditis
Treatment references
1. Loss KL, Shaddy RE, Kantor PF: Recent and Upcoming Drug Therapies for Pediatric Heart Failure. Front Pediatr 9:681224, 2021. Published 2021 Nov 11. doi:10.3389/fped.2021.681224
2. Oster ME, Kelleman M, McCracken C, et al: Association of digoxin with interstage mortality: Results from the Pediatric Heart Network Single Ventricle Reconstruction Trial Public Use Dataset. J Am Heart Assoc 5(1): e002566., 2016.
3. Bolin EH, Lang SM, Tang X, et al: Propranolol versus digoxin in the neonate for supraventricular tachycardia (from the Pediatric Health Information System). Am J Cardiol 119(10): 1605–1610, 2017.
4. Baltimore RS, Gewitz M, Baddour LM, et al: Infective Endocarditis in Childhood: 2015 Update: A Scientific Statement From the American Heart Association. Circulation 132(15):1487–1515, 2015. doi:10.1161/CIR.0000000000000298
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
American Heart Association: Common Heart Defects: Provides overview of common congenital heart defects for parents and caregivers
American Heart Association: Infective Endocarditis: Provides an overview of infective endocarditis, including summarizing prophylactic antibiotic use, for patients and caregivers






