




Le sindromi di Ehlers-Danlos sono malattie ereditarie del collagene caratterizzata da ipermobilità articolare, iperlassità del derma e fragilità diffusa dei tessuti. La diagnosi è clinica. Il trattamento è di supporto.
L'eredità è solitamente autosomica dominante, ma le sindromi di Ehlers-Danlos sono eterogenee. Mutazioni di geni differenti interessano il quantitativo, la struttura, o l'assemblaggio delle varie forme di collagene. Possono esistere mutazioni nei geni che codificano diversi tipi di collagene (p. es., tipo I, III o V) o enzimi che modificano il collagene (p. es., lisil idrossilasi, una proteasi che scinde il collagene).
Esistono 6 tipi principali:
Ipermobile
Classico
Vascolare
Cifoscoliosi
Artrocalasia
Dermatosparassi
I tipi ipermobili, classici e vascolari sono i più frequenti.
Ci sono anche 7 tipi rari.
Sintomatologia delle sindromi di Ehlers-Danlos
I sintomi e i segni delle sindromi di Ehlers-Danlos variano ampiamente, ma alcune manifestazioni sono considerate caratteristiche di diversi tipi.
I sintomi predominanti comprendono ipermobilità articolare, anomalia nella formazione di cicatrici e nella guarigione delle ferite, fragilità vascolare, e cute vellutata, iperestensibile. La cute può essere stirata per parecchi centimetri, ma una volta rilasciata, ritorna nella posizione normale.
Spesso sono presenti delle grosse cicatrici a carta di papiro sulle prominenze ossee, in particolare sui gomiti, sulle ginocchia e sulle tibie; la cicatrizzazione è meno grave nel tipo ipermobile. Sopra le cicatrici, o nelle zone su cui si esercita una maggiore pressione, si formano spesso pseudotumori molluscoidi (protuberanze cutanee).
L'entità dell'ipermobilità articolare può essere variabile, ma può essere marcata nelle forme classica, artrocalasia e ipermobile.
La tendenza al sanguinamento è rara, benché il tipo vascolare sia caratterizzato da rottura vascolare ed ecchimosi.
Si possono palpare o osservare sulle RX delle sferule calcifiche sottocutanee.

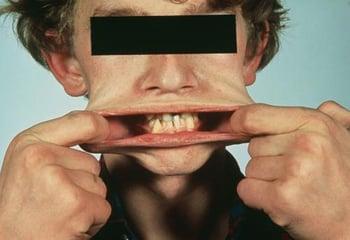
Complicazioni delle sindromi di Ehlers-Danlos
Traumi lievi possono causare ferite ampie e beanti, ma poco sanguinanti; la sutura delle ferite chirurgiche può essere difficile poiché i punti di sutura tendono a strapparsi facilmente dal tessuto fragile. Le complicanze chirurgiche si verificano a causa della fragilità dei tessuti profondi.
La sclera può essere fragile, portando alla perforazione del bulbo oculare nel tipo cifoscoliotico.
Spesso compaiono modesti versamenti articolari, distorsioni e lussazioni. La cifoscoliosi vertebrale è presente nel 25% dei pazienti (specialmente in quelli con il tipo cifoscoliotico), una deformità toracica nel 20% e un piede equino-varo-supinato nel 5%. Circa il 90% degli adulti colpiti ha il piede piatto. La displasia dello sviluppo dell'anca (conosciuta anche come lussazione congenita dell'anca) si verifica nell'1% (il tipo artrocalasi è caratterizzato da displasia bilaterale dello sviluppo dell'anca).
Sono frequenti ernie e diverticoli gastrointestinali. Raramente, porzioni del tratto gastrointestinale sanguinano o si perforano spontaneamente, mentre gli aneurismi aortici dissecanti e le grosse arterie vanno incontro a rottura spontanea.
Il prolasso valvolare è una complicanza frequente nel tipo più grave (tipo vascolare).
Nelle donne in gravidanza, l'estensibilità tissutale può causare parto prematuro, incompetenza cervicale, ed eventualmente rottura uterina; se il feto è affetto, la membrana fetale è fragile, causando talvolta la rottura precoce. La fragilità dei tessuti materni può complicare l'episiotomia o un parto cesareo. Si possono avere emorragie pre-, peri- e post-natali.
Altre potenziali gravi complicanze comprendono fistola arterovenosa, rottura di un viscere e pneumotorace o pneumoemotorace.
Diagnosi delle sindromi di Ehlers-Danlos
Criteri clinici
Test genetici di conferma
Ecocardiografia e/o un altro imaging vascolare per cercare complicanze cardiovascolari
La maggior parte, ma non tutti i tipi di sindrome di Ehlers-Danlos, coinvolge uno o entrambi dei seguenti:
Ipermobilità articolare
Iperestensibilità articolare
Così, la diagnosi delle sindromi di Ehlers-Danlos può essere sospettata in pazienti che si presentano con disturbi come lussazioni articolari frequenti, scarsa guarigione delle ferite e/o cicatrici frequenti o inusuali. Tuttavia, l'ipermobilità articolare è frequente tra la popolazione generale. Inoltre, altre patologie del tessuto connettivo con simili manifestazioni articolari e/o cutanee (p. es., sindrome di Marfan, cutis laxa) devono essere considerate.
Lo screening dei pazienti con semplici domande riguardanti l'ipermobilità articolare può essere utile (1):
Riesci ora, o sei riuscito in passato, a posare le mani sul pavimento senza piegare le ginocchia?
Riesci ora, o sei riuscito in passato, a piegare il pollice per toccare l'avambraccio?
Da bambino, ti sei divertito con i tuoi amici contorcendo il tuo corpo in forme strane o potresti fare la spaccata?
Da bambino o adolescente, le ginocchia o le spalle si sono lussate in più di un'occasione?
Considera di avere "doppie articolazioni"?
Una risposta positiva a 2 o più di queste domande suggerisce fortemente ipermobilità, che può essere ulteriormente valutata con l'esame obiettivo utilizzando il Beighton scoring system. Questo strumento valuta l'ipermobilità in 4 paia di articolazioni (5e dita, pollici, gomiti, ginocchia) e colonna vertebrale. Viene dato un punto per ogni articolazione che manifesta il criterio di ipermobilità definito; un punteggio ≥ 5 è considerato indicare ipermobilità (2).
L'iperestensibilità cutanea è valutata in aree standardizzate. La cute è considerata iperestensibile se può essere estesa > 1,5 cm sull'avambraccio distale e sul dorso delle mani, > 3 cm sul collo, gomito e ginocchia e > 1 cm sul palmo (2).
Ci sono criteri clinici maggiori e minori per ciascun tipo di Ehlers-Danlos (2), che comprendono tipicamente la presenza o l'assenza di ipermobilità articolare e di iperestensibilità cutanea. Tuttavia, vi è molta variabilità sia all'interno che tra i diversi tipi, e la diagnosi deve essere confermata da test genetici, ora disponibili per la maggior parte dei sottotipi.
L'esame ultrastrutturale di una biopsia cutanea può contribuire a diagnosticare i tipi classico, ipermobile e vascolare.
L'ecocardiografia e altre imaging vascolari sono eseguite per ricercare disturbi cardiaci (p. es., prolasso valvolare, aneurisma arterioso) che sono associati ad alcuni tipi.
Riferimenti relativi alla diagnosi
1. Hakim AJ, Grahame R: A simple questionnaire to detect hypermobility: An adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract 57(3):163–166, 2003. PMID: 12723715
2. The Ehlers-Danlos Society: EDS types: 2017 International Diagnostic Criteria. Consultato il 11/10/2022.
Prognosi delle sindromi di Ehlers-Danlos
L'aspettativa di vita in genere è normale con la maggior parte dei tipi.
Complicanze potenzialmente letali si verificano in alcuni tipi (p. es., rottura arteriosa nel tipo vascolare).
Trattamento delle sindromi di Ehlers-Danlos
Prima identificazione e trattamento delle complicanze
Non esiste una terapia specifica per le sindromi di Ehlers-Danlos.
Occorre ridurre al minimo i traumi. L'uso di indumenti protettivi o imbottiti può essere d'aiuto.
In caso di interventi chirurgici l'emostasi deve essere meticolosa. Le ferite vanno suturate attentamente evitando trazioni eccessive sui tessuti.
È obbligatoria la supervisione ostetrica durante la gravidanza e il parto. Occorre fornire una consulenza genetica.
Punti chiave
Le sindromi di Ehlers-Danlos sono malattie genetiche del tessuto connettivo con 13 tipi clinici identificati.
I pazienti presentano tipicamente articolazioni ipermobili, cute iperestensibile e/o cute e tessuto connettivo fragili.
Il tessuto connettivo fragile può predisporre a rigurgito valvolare cardiaco, rottura sclerale, dissezione o rottura arteriosa, rottura uterina e altre complicanze.
La diagnosi viene effettuata utilizzando criteri clinici e in genere confermata da test genetici.
Per ulteriori informazioni
Le seguenti risorse in lingua inglese possono essere utili. Si noti che il Manuale non è responsabile per il contenuto di queste risorse.
Beighton scoring system for assessing joint hypermobility in Ehlers-Danlos syndrome
American Journal of Medical Genetics Part C: Seminars in Medical Genetics: The 2017 EDS Classification







