




L'osteogenesi imperfetta è una malattia ereditaria del collagene che causa una diffusa anormale fragilità delle ossa e a volte è associata ad ipoacusia neurosensoriale, sclere blu, dentinogenesi imperfetta e ipermobilità articolare. La diagnosi è generalmente clinica. Il trattamento comprende l'ormone della crescita per alcuni tipii bifosfonati e il denosumab.
Esistono 4 tipi principali di osteogenesi imperfetta (1):
I: Mild, sclere blu
II: Neonatale letale, sclere blu
III: Colorazione progressiva, sclerale variabile
IV: variabile e deformante, colore sclerale normale
L'ereditarietà è solitamente di tipo autosomico dominante. Il 90% delle persone che presentano uno dei tipi principali ha mutazioni nei geni che codificano le catene pro-alfa del procollagene di tipo I (una componente strutturale di ossa, legamenti e tendini), COL1A1 o COL1A2.
Ci sono un certo numero di altri tipi più rari (tipi da V a XXI), che sono causati da mutazioni in geni diversi.
Riferimento
1. Van Dijk FS, Sillence DO: Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164A(6):1470–1481, 2014. doi: 10.1002/ajmg.a.36545. Clarification and additional information. Am J Med Genet A 167A(5):1178, 2015. doi: 10.1002/ajmg.a.36784
Sintomatologia dell'osteogenesi imperfetta
La perdita dell'udito è presente nel 50-65% di tutti i pazienti affetti da osteogenesi imperfetta e può comparire in tutti i 4 tipi principali.
Il tipo I è il meno grave. La sintomatologia in alcuni pazienti è limitata alle sclere blu (dovute a un deficit nel tessuto connettivo che permette ai vasi sottostanti di essere visibili in trasparenza) e al dolore muscoloscheletrico da ipermobilità articolare. Sono possibili fratture ricorrenti nell'infanzia.
Il tipo II (tipo letale neonatale o osteogenesi imperfetta congenita) è il più grave ed è letale. Le fratture congenite multiple portano a un accorciamento degli arti. Le sclere sono blu. Il cranio è molle, quando si palpa sembra un "sacco pieno d'ossa". Poiché il cranio è molle, un trauma durante il parto può causare emorragia endocranica e natimortalità, oppure i neonati possono morire all'improvviso durante i primi giorni o le prime settimane di vita.

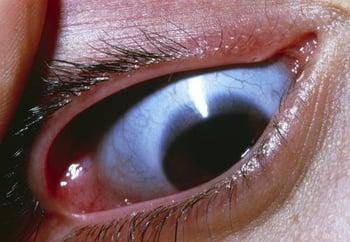
Il tipo III è progressivo e la forma più grave non letale di osteogenesi imperfetta. I pazienti affetti dal tipo III hanno bassa statura, curvatura della colonna vertebrale, e fratture multiple, ricorrenti. Macrocefalia con facies triangolare e deformità del petto sono frequenti. La tonalità delle sclere varia.


Il tipo IV è di gravità intermedia. Il tasso di sopravvivenza è elevato. Questo tipo è variabile e deformante. Le ossa si fratturano facilmente nell'infanzia prima dell'adolescenza. Le sclere sono tipicamente di colore normale. L'altezza è moderata-bassa. Una diagnosi accurata è importante poiché questi pazienti possono trarre beneficio dal trattamento.
Diagnosi dell'osteogenesi imperfetta
Valutazione clinica
Talvolta analisi del procollagene di tipo I o test genetici
La diagnosi di osteogenesi imperfetta solitamente è clinica, ma non esistono criteri standardizzati.
L'analisi del procollagene del tipo I da colture di fibroblasti (da una biopsia cutanea) o l'analisi di sequenza dei geni COL1A1 e COL1A2 possono essere usate quando la diagnosi clinica non è chiara.
Una grave osteogenesi imperfetta può essere rilevata in utero con ecografia di livello II.
Trattamento dell'osteogenesi imperfetta
Ormone della crescita
Bifosfonati
A volte denosumab
A volte vitamina D
L'ormone della crescita aiuta i bambini che ad esso rispondono (tipo I e IV).
Il trattamento con bifosfonati ha lo scopo di aumentare la densità ossea e ridurre il dolore osseo e il rischio di fratture (1). Si usa pamidronato EV (da 0,5 a 3 mg/kg 1 volta/die per 3 giorni, ripetuto secondo necessità ogni 4-6 mesi) o alendronato orale (1 mg/kg, 20 mg massimo, 1 volta/die).
Il denosumab è un potente inibitore del riassorbimento osteoclastico osseo ed è tipicamente somministrato in iniezione. Studi hanno dimostrato che questo farmaco è utile in alcuni pazienti con artrosi (2).
La supplementazione di vitamina D deve essere fornita alle persone che sono carenti di questo ormone.
Chirurgia ortopedica, fisioterapia, e terapia occupazionale aiutano a prevenire le fratture e a migliorare la funzione.
L'impianto cocleare è indicato in casi selezionati di perdita dell'udito.
Riferimenti relativi al trattamento
1. Dwan K, Phillipi CA, Steiner RD, Basel D: Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev CD005088, 2016. doi: 10.1002/14651858.CD005088.pub4
2. Li G, Jin Y, Levine MAH, et al: Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr 107(3):534–537, 2018. doi: 10.1111/apa.14154
Per ulteriori informazioni
Le seguenti risorse in lingua inglese possono essere utili. Si noti che il Manuale non è responsabile per il contenuto di questa risorsa.
Osteogenesis Imperfecta (OI) Foundation: An organization providing support, education, and research information about OI







