


Die eosinophile Granulomatose mit Polyangiitis ist eine systemische, kleine und mittlere Gefäße nekrotisierende Vaskulitis, charakterisiert durch extravaskuläre Granulome, Eosinophilie und Gewebeinfiltration infolge Eosinophile. Sie tritt bei Menschen mit Asthma im Erwachsenenalter, allergischer Rhinitis, Nasenpolypen oder einer Kombination auf. Die Diagnose wird am besten durch Biopsie bestätigt. Die Behandlung erfolgt in erster Linie mit Kortikosteroiden und einem weiteren Immunsuppressivum.
(Siehe auch Übersicht über Vaskulitis.)
Eosinophile Granulomatose mit Polyangiitis (EGPA) kommt bei etwa 3 auf eine Millionen Menschen vor. das Durchschnittsalter bei Krankheitsbeginn liegt bei 48 Jahren.
EGPA ist gekennzeichnet durch nekrotisierende extravaskuläre Granulome (in der Regel reich an Eosinophilen), Eosinophilie und Gewebeinfiltration durch Eosinophile. Allerdings bestehen diese Anomalien nicht immer gleichzeitig. Die Vaskulitis betrifft typischerweise kleine und mittlere Arterien. Jedes Organ kann betroffen sein, aber Lunge, Haut, Nase, Herz-Kreislauf-System, Nieren, peripheres Nervensystems, zentrales Nervensystem, Gelenke und Gastrointestinaltrakt sind am häufigsten betroffen. Selten kann eine pulmonale Kapillaritis zu einer alveolären Hämorrhagie führen.
Ätiologie von EGPA
Die Ursache der eosinophilen Granulomatose mit Polyangiitis ist unbekannt. Allerdings ist ein allergischer Mechanismus beteiligt, bei dem das Gewebe direkt durch Eosinophile und durch Degranulationsprodukte von Neutrophilen geschädigt wird. Die Aktivierung von T-Lymphozyten scheint zur Aufrechterhaltung der eosinophilen Entzündung beizutragen. Das Syndrom tritt auf bei Patienten, die Asthma mit Beginn im Erwachsenenalter, allergische Rhinitis, Nasenpolypen oder eine Kombination davon zeigen. Antineutrophile zytoplasmatische Autoantikörper (ANCA) sind in 40% der Fälle vorhanden.
Symptome und Anzeichen von EGPA
Das Syndrom hat drei Phasen, die sich überlappen können:
Prodromal: Diese Phase kann über Jahre bestehen bleiben. Die Patienten haben allergische Rhinitis, Nasenpolypen, Asthma oder eine Kombination.
2. Phase: Eosinophilie im peripheren Blut und Gewebe ist typisch. Zur klinischen Präsentation, die dem Löffler-Syndrom ähnelt, gehören chronische eosinophile Pneumonie und eosinophile Gastroenteritis.
3. Phase: Es entwickelt sich eine potenziell lebensbedrohliche Vaskulitis. Organfunktionsstörungen und systemische Symptome (z. B. Fieber, Unwohlsein, Gewichtsverlust, Müdigkeit) sind in dieser Phase häufig.
Allerdings folgen die Phasen nicht notwendigerweise aufeinander, und das Zeitintervall zwischen ihnen variiert stark.
Verschiedene Organe und Systeme sind betroffen:
Atemwege: Asthma, oft mit Beginn im Erwachsenenalter, tritt bei den meisten Patienten auf und tendiert zu schwerem und kortikosteroidabhängigem Verlauf. Sinusitis ist häufig, aber nicht destruktiv, ohne schwere nekrotisierende Entzündung. Die Patienten sind kurzatmig. Transiente fleckige Lungeninfiltrate sind häufig.
Neurologisch: Neurologische Symptome sind sehr häufig. Multiple Mononeuropathie (Mononeuritis multiplex) tritt bei bis zu drei Viertel der Patienten auf. Eine Beteiligung des zentralen Nervensystems ist selten, kann aber Hemiparese, Verwirrung, Krampfanfälle und Koma mit oder ohne Hirnnervenlähmungen oder Anzeichen eines Hirninfarkts umfassen.
Kutan: Die Haut ist bei etwa die Hälfte der Patienten betroffen. Knötchen und Papeln zeigen sich auf den Streckseiten der Extremitäten. Sie werden durch extravaskuläre palisading granulomatöse Läsionen mit zentraler Nekrose verursacht. Es können sich Purpura oder erythematöse Papeln aufgrund leukozytoklastischer Vaskulitis mit oder ohne prominente eosinophile Infiltration entwickeln.
Skelettmuskulatur: Arthralgien, Myalgien oder sogar Arthritis können auftreten.
Herz: Die kardiale Beteiligung, eine der Hauptursachen für die Sterblichkeit, umfasst Herzinsuffizienz aufgrund von Myokarditis und endomyokardialer Fibrose, koronare Vaskulitis (eventuell mit Myokardinfarkt), Herzklappenerkrankungen und Perikarditis. Der vorherrschende histopathologische Befund ist eine eosinophile Myokarditis.
Gastrointestinal: Bis zu einem Drittel der Patienten zeigt gastrointestinale Symptome (z. B. Bauchschmerzen, Diarrhö, Blutungen, akalkulöse Cholezystitis) aufgrund einer eosinophilen Gastroenteritis oder eine Mesenterialischämie aufgrund einer Vaskulitis.
Renal: Die Nieren sind weniger häufig betroffen als bei anderen vaskulitischen Erkrankungen, die mit ANCA (antineutrophil cytoplasmic autoantibodies) assoziiert sind. Typischerweise liegt eine pauci-immune (mit wenn überhaupt nur wenigen Immunkomplexen), fokal segmentale, nekrotisierende Glomerulonephritis mit Halbmondbildung vor; eine eosinophile oder granulomatöse Entzündung der Nieren ist selten.

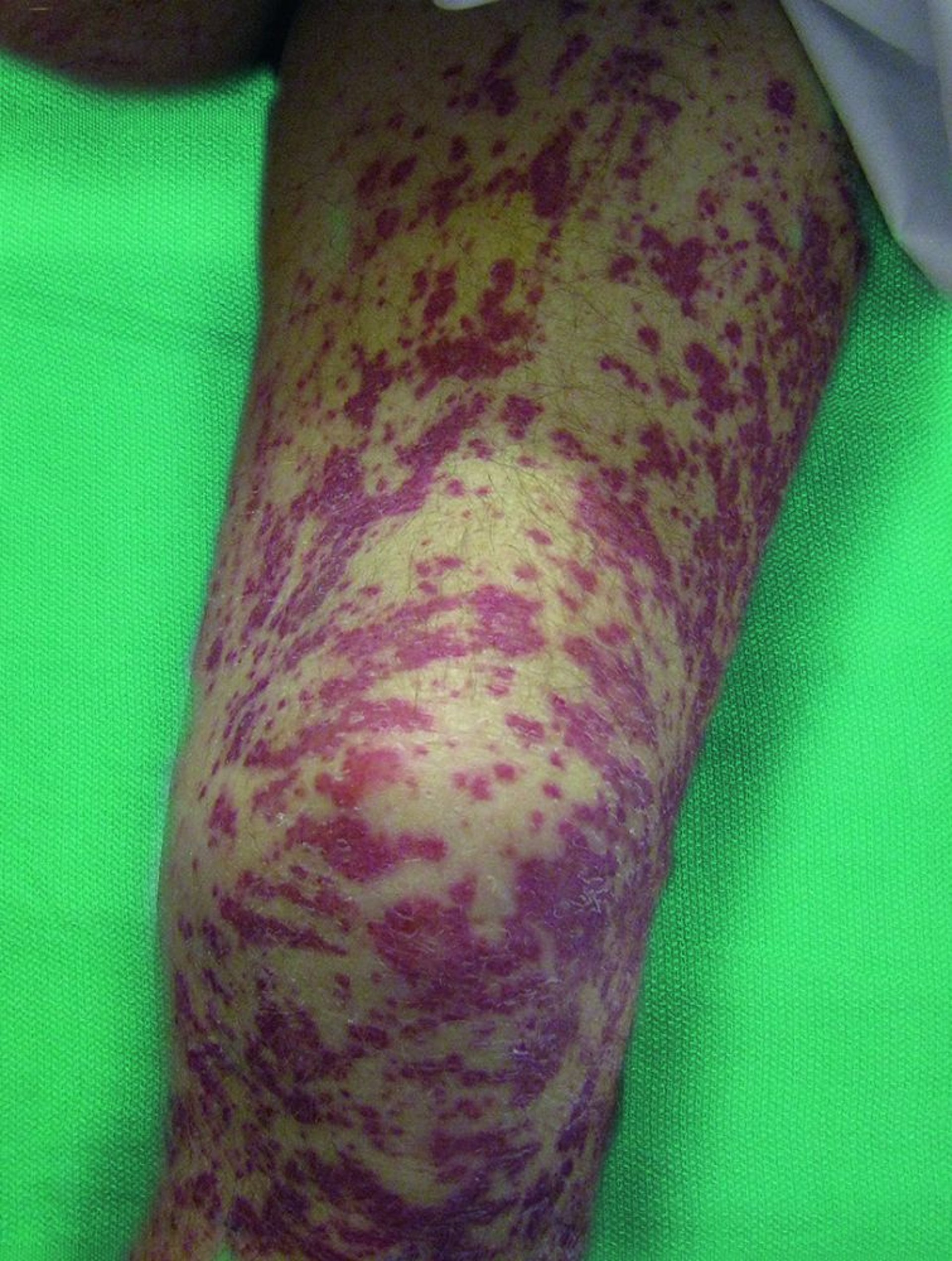
© Springer Science+Business Media
Eine renale, kardiale oder neurologische Beteiligung bedeutet eine schlechtere Prognose.
Diagnose von EGPA
Klinische Kriterien
Routinelaboruntersuchungen
Echokardiographie
Biopsie
Die Chapel Hill Consensus Conference von (1) 2012 definiert die eosinophilen Granulomatose mit Polyangiitis als eine an Eosinophilen reiche und nekrotisierende granulomatöse Entzündung unter Beteiligung der Atemwege mit nekrotisierender Vaskulitis der kleinen und mittelgroßen Gefäße in Verbindung mit Asthma und Eosinophilie. Criteria for classification from the American College of Rheumatology consist of the following:
Asthma
Eosinophilie von > 10% im peripheren Blut
Nasennebenhöhlenentzündung
Lungeninfiltrate, manchmal vorübergehend
Histologischer Nachweis einer Vaskulitis mit extravaskulären Eosinophilen
Mehrere Mononeuropathien oder Polyneuropathie
Wenn ≥ 4 Kriterien vorhanden sind, beträgt die Sensitivität 85% und die Spezifität 99,7%.
Die Tests zielen darauf ab, die Diagnose und das Ausmaß der Organbeteiligung zu sichern und die EGPA von anderen eosinophilen Erkrankungen (z. B. parasitäre Infektionen, Arzneimittelwirkungen, akute und chronische eosinophile Pneumonie, und chronische eosinophile Pneumonie, allergische bronchopulmonale Aspergillose, hypereosinophiles Syndrom) abzugrenzen. Die Diagnose von EGPA wird durch klinische Befunde und Ergebnisse der routinemäßigen Labortests gestellt, sollte aber in der Regel durch eine Biopsie der Lunge oder anderer betroffener Gewebe bestätigt werden.
Blutuntersuchungen und Röntgenaufnahmen des Thorax werden angefertigt, die Ergebnisse sind jedoch nicht diagnostisch. Ein Differenzialblutbild wird erstellt, um auf Eosinophilie, die auch bei einigen Patienten ein Marker für die Krankheitsaktivität ist, zu prüfen. IgE- und CRP-Spiegel sowie die Erythrozytensedimentationsrate werden in regelmäßigen Abständen bestimmt, um die inflammatorische Aktivität zu beurteilen. Urinanalyse und Kreatininbestimmung werden durchgeführt, um auf Nierenerkrankungen zu screenen und den Schweregrad zu überwachen. Die Elektrolytspiegel werden gemessen.
Bei serologischen Tests werden bei bis zu 40% der Patienten antineutrophile zytoplasmatische Autoantikörper (ANCA) gefunden; in diesem Fall wird ein Enzyme-linked immunosorbent assay (ELISA) durchgeführt, um auf spezifische Antikörper zu testen. Perinukleäre ANCA (p-ANCA) mit Antikörpern gegen Myeloperoxidase ist ein sehr häufiges Ergebnis, jedoch ist ANCA kein spezifischer oder sensitiver Test für EGPA.
Obwohl als Marker für die Krankheitsaktivität verwendet, erreichen Eosinophilie, IgE, ANCA, Erythrozytensedimentationsrate und C-reaktive-Proteinspiegel dies und sagen Flare-ups nur mit erheblichen Einschränkungen voraus.
Der Röntgenthorax zeigt oft transiente fleckförmige Lungeninfiltrate.
Ein 2D-Echokardiogramm des Herzens sollte bei allen Patienten zu Beginn der Studie erhoben und bei Auftreten von Symptomen und/oder Anzeichen einer Herzinsuffizienz wiederholt werden.
Wenn möglich, sollte eine Biopsie des am besten zugänglichen betroffenen Gewebes erfolgen.
Diagnosehinweis
1. Jennette JC, Falk RJ, Bacon PA, et al: 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65(1):1-11, 2013. doi: 10.1002/art.37715
Behandlung von EGPA
Kortikosteroide
Systemische Kortikosteroide in Kombination mit einem Immunsuppressivum sind die Hauptstütze der Behandlung der eosinophilen Granulomatose mit Polyangiitis (EGPA), da Kortikosteroide allein oft keine Remission aufrechterhalten können, selbst wenn keine schlechten prognostischen Faktoren vorliegen. Andere Immunsuppressiva (z. B. Cyclophosphamid, Rituximab, Methotrexat, Azathioprin) werden hinzugefügt, wobei die Wahl der Mittel vom Schweregrad und der Art der Organbeteiligung abhängt und dieselben allgemeinen Kriterien für die Behandlung der Granulomatose mit Polyangiitis oder der mikroskopischen Polyangiitis gelten. In einer retrospektiven Studie von 41 Patienten mit EGPA, die mit Rituximab behandelt wurden, waren 49% nach 12 Monaten in der Remissionsphase und Rituximab verringerte den Bedarf an Kortikosteroiden. Diese Ergebnisse schneiden im Vergleich zu anderen Behandlungen positiv ab. Mepolizumab, 300 mg subkutan alle 4 Wochen, senkt nachweislich die Rückfallquote bei leichter bis mittelschwerer, schubförmiger oder refraktärer EGPA. Der Nutzen lag vor allem bei den oberen und unteren Atemwegen, einschließlich aktivem Asthma (1). Die Wirkung von Mepolizumab bei Patienten mit schweren Manifestationen, einschließlich kardialer Erkrankungen, ist unbekannt.
Eine Herzbeteiligung in Form einer Myokarditis mit Herzversagen ist die Haupttodesursache bei EGPA.
Literatur zur Therapie
Wechsler ME, Akuthota P, Jayne D, et al: Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med 376(20):1921-1932, 2017. doi:10.1056/NEJMoa1702079
Wichtige Punkte
Bei der eosinophilen Granulomatose mit Polyangiitis handelt es sich um eine seltene, kleine und mittelgroße Gefäße betreffende Vaskulitis.
Die Phasen umfassen Symptome der oberen Atemwege und Atemnot, eosinophile Pneumonie und Gastroenteritis sowie eine lebensbedrohliche Vaskulitis.
Die Phasen treten in oder außerhalb der Reihenfolge auf und überschneiden sich.
Eine kardiale oder neurologische Beteiligung kann auftreten und deutet auf eine schlechte Prognose hin.
Die Diagnose wird anhand klinischer Kriterien, durch routinemäßige Laboruntersuchungen und manchmal eine Biopsie gestellt.
Führen Sie bei allen Patienten ein 2D-Echokardiogramm durch.
Die Behandlung erfolgt mit Kortikosteroiden und Immunsuppressiva, basierend auf dem Schweregrad der Erkrankung. Wobei für die Behandlung von Granulomatose mit Polyangiitis oder mikroskopischer Polyangiitis die gleichen Kriterien gelten.
Erwägen Sie die Behandlung mit Rituximab wegen der möglichen hohen Ansprechraten und des reduzierten Bedarfs an Kortikosteroiden.
Erwägen Sie Mepolizumab bei rezidivierender oder refraktärer Erkrankung mit primär oberen und/oder unteren Atemwegsmanifestationen.
Weitere Informationen
Die folgenden englischsprachigen Quellen können nützlich sein. Bitte beachten Sie, dass das MSD-Manual nicht für den Inhalt dieser Quellen verantwortlich ist.
American College of Rheumatology (ACR): Listet eine Reihe von ACR-genehmigten Klassifikationskriterien-Sets auf und bietet Informationen über den Zweck von Kriterien-Sets, ihre Entwicklung und Validierung und die Rolle des ACR bei der Annahme dieser Kriterien.