


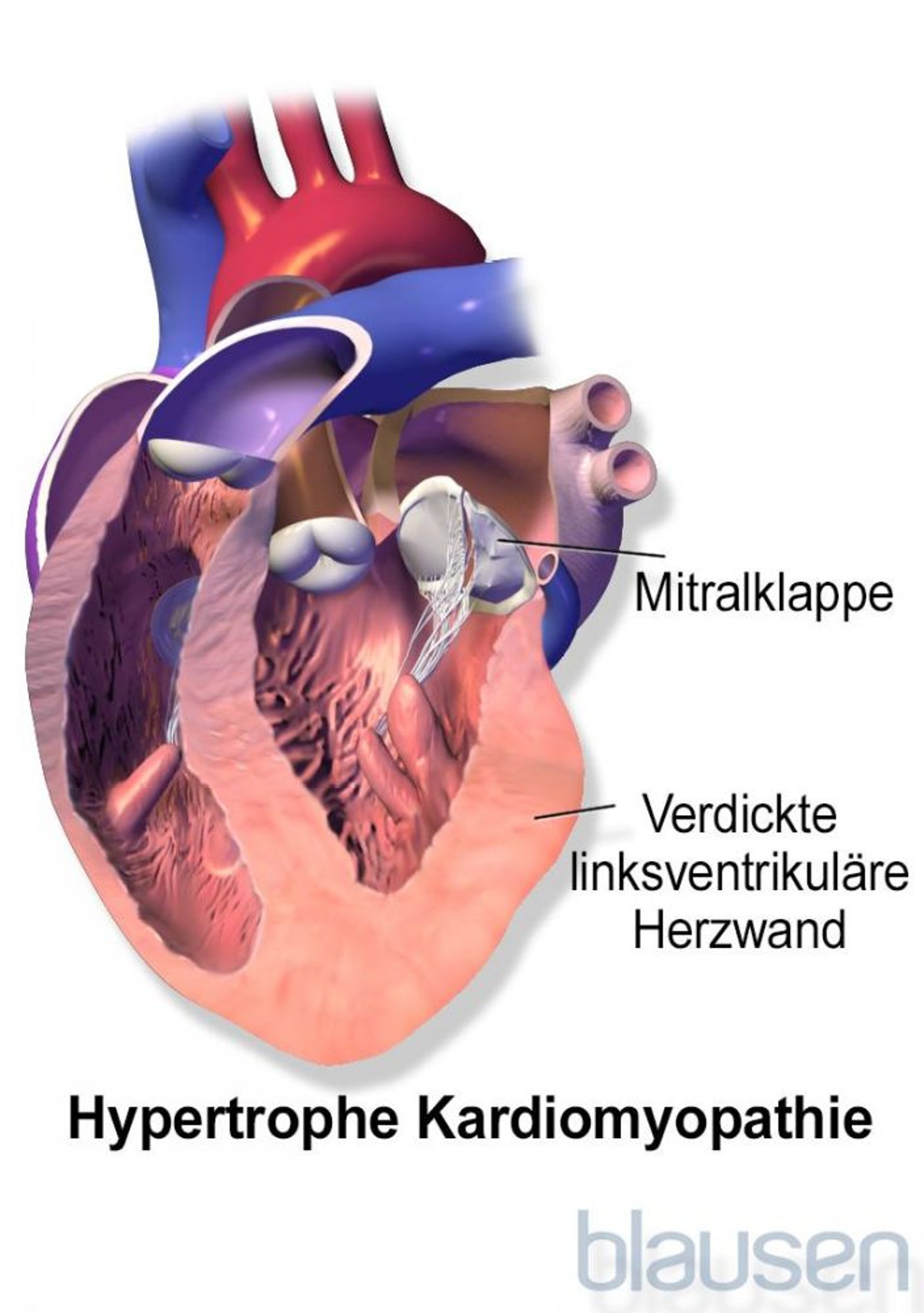
Zur hypertrophen Kardiomyopathie zählen Herzkrankheiten, bei denen sich die Wände der Ventrikel (die zwei unteren Herzkammern) verdicken (Hypertrophie) und unelastisch werden.
Die meisten Fälle von hypertropher Kardiomyopathie sind auf einen erblich bedingten, genetischen Defekt zurückzuführen.
Bei den Betroffenen kommt es zu Ohnmacht, Schmerzen im Brustbereich, Kurzatmigkeit und Palpitationen (Wahrnehmung von unregelmäßigen Herzschlägen).
Die Diagnose wird anfänglich auf der Grundlage einer körperlichen Untersuchung gestellt und dann mittels Echokardiografie oder Magnetresonanztomografie bestätigt.
Die Behandlung erfolgt mit Medikamenten, die die Stärke der Herzkontraktionen verringern.
Bei Kardiomyopathie werden Struktur und Funktion der Muskelwände der Herzkammern zunehmend beeinträchtigt. Es gibt drei Hauptarten der Kardiomyopathie. Zusätzlich zur hypertrophen Kardiomyopathie gibt es noch die kongestive Kardiomyopathie und die restriktive Kardiomyopathie (siehe auch Übersicht über Kardiomyopathie).
Der Begriff Kardiomyopathie wird nur für eine Krankheit verwendet, die den Herzmuskel direkt betrifft. Andere Erkrankungen wie Bluthochdruck und anormale Herzklappen (z. B. Aortenstenose) können letztendlich ebenfalls zu einem verdickten Herzmuskel und einer Herzinsuffizienz führen.
Die hypertrophe Kardiomyopathie ist eine häufige Ursache für plötzlichen Tod bei jungen Athleten. Mindestens eine von 500 Personen ist betroffen.
Ursachen einer hypertrophen Kardiomyopathie
Die hypertrophe Kardiomyopathie beruht fast immer auf einem Gendefekt. Dabei kann es sich um Folgendes handeln:
spontane Genmutation
vererbter Gendefekt
Eine hypertrophe Kardiomyopathie ist sehr selten bei Menschen mit Erkrankungen wie Akromegalie (übermäßigem Wachstum aufgrund einer Überproduktion des Wachstumshormons, gewöhnlich durch einen gutartigen Hypophysentumor), einem Phäochromozytom (Tumor, der das Hormon Epinephrin überproduziert) oder einer Neurofibromatose (einer genetischen Erkrankung, bei der es zu unkontrolliertem Wachstum von Nervengewebe unter der Haut und in anderen Teilen des Körpers kommt).

Komplikationen
Die dicken, steifen Wände der Ventrikel entspannen sich nicht ausreichend, damit sich die Herzkammern mit Blut füllen können. Dieses Problem verstärkt sich, wenn das Herz schnell schlägt (wie beispielsweise beim Sport), weil das Herz dann noch weniger Zeit hat, sich mit Blut zu füllen. Wenn sich das Herz nicht richtig füllt, pumpt es mit jedem Schlag weniger Blut. Manchmal beeinträchtigen die verdickten Herzwände auch den Blutstrom aus dem Herzen heraus. Bei dieser Variante spricht man von hypertropher obstruktiver Kardiomyopathie.
Da sich die Ventrikelwände verdicken, kann sich die Mitralklappe (die Klappe, die sich aus dem linken Vorhof in den linken Ventrikel öffnet) nicht mehr komplett schließen, sodass Blut in den linken Vorhof zurückfließt (Mitralinsuffizienz). Eine solche undichte Herzklappe in Verbindung mit verdickten Ventrikelwänden verursacht typischerweise abnorme Herzgeräusche.
Herzrhythmusstörungen können auftreten, was zum plötzlichen Tod führen kann.
Symptome einer hypertrophen Kardiomyopathie
Die möglicherweise auftretenden Symptome sind sehr unterschiedlich, entwickeln sich jedoch gewöhnlich zwischen dem 20. und 40. Lebensjahr. Symptome, die zuerst während einer Belastung auftreten, umfassen u. a. Folgendes:
Ohnmachtsanfall (Synkope)
Schmerzen im Brustkorb
Kurzatmigkeit
Empfinden unregelmäßiger Herzschläge (Palpitationen)
Eine Ohnmacht tritt gewöhnlich plötzlich und ohne Warnsignale auf. Ohnmacht oder sogar plötzlicher Tod können die ersten Anzeichen für diese Erkrankung sein.
Diagnose einer hypertrophen Kardiomyopathie
Echokardiografie und/oder Magnetresonanztomografie (MRT) des Herzens
Die anfängliche Diagnose einer hypertrophen Kardiomyopathie beruht in der Regel auf den beobachteten Symptomen sowie auf den Ergebnissen einer körperlichen Untersuchung, einer Elektrokardiografie (EKG) und einer Röntgenuntersuchung des Brustkorbs. Ebenso können mit einem Stethoskop abgehörte Herzgeräusche die Diagnose unterstützen. Obwohl eine EKG und Echokardiografie nicht für alle jungen Leistungssportler empfohlen werden, sollten alle Sportler in diesem Bereich auf Risikofaktoren hin untersucht werden, die weitere Untersuchungen erforderlich machen.
Die Diagnose kann am besten mit einer Echokardiografie bestätigt werden, jedoch kann auch eine MRT des Herzens verwendet werden, da diese genauere Informationen liefern kann.
Die Herzkatheterisierung ist ein invasiver Eingriff, bei dem ein Katheter über ein Blutgefäß im Arm, Hals oder Bein in das Herz eingeführt wird. Sie wird oft durchgeführt, um den Druck in den Herzkammern und den Schweregrad der Blockierung des Blutflusses aus der linken Herzkammer aufgrund der verdickten Wände zu messen.
Da eine hypertrophe Kardiomyopathie in der Regel von einer Genmutation verursacht wird, können auch genetische Untersuchungen durchgeführt werden, um festzustellen, ob Verwandte betroffen sind.
Prognose bei einer hypertrophen Kardiomyopathie
Etwa ein Prozent der Erwachsenen mit hypertropher Kardiomyopathie stirbt jedes Jahr. Bei Kindern mit hypertropher Kardiomyopathie ist die Sterblichkeit höher als bei Erwachsenen.
Zu den anderen Faktoren, die ein erhöhtes Sterberisiko nahelegen, gehören u. a:
Plötzlicher Tod in der Familie, insbesondere plötzlicher frühzeitiger Tod
Unerklärlicher Ohnmachtsanfall, Herzstillstand oder ventrikuläre Tachykardie
Auftreten eines rasenden oder Herzrhythmusstörungen
Schweregrad der Verdickung im Herzmuskel
Unfähigkeit des Herzens, ausreichend Blut zu pumpen
Charakteristische MRT-Befunde
Der Tod tritt gewöhnlich plötzlich ein, vermutlich aufgrund von Herzrhythmusstörungen. Seltener führt eine chronische Herzinsuffizienz zum Tod.
Menschen, die mit dieser Erkrankung geboren wurden, sollten sich bei der Familienplanung genetisch beraten lassen, weil die Wahrscheinlichkeit der Weitervererbung dieser Krankheit 50 % beträgt. Familienangehörige ersten Grades (Geschwister oder Kinder) von Patienten mit dieser Erbkrankheit sollten ihr Herz untersuchen lassen (entweder mit einem genetischen Test und/oder einer routinemäßigen Echokardiografie).
Behandlung einer hypertrophen Kardiomyopathie
Arzneimittel wie Betablocker und/oder Kalziumkanalblocker
Manchmal ein Verfahren zur Verbesserung des Blutflusses
Manchmal ein implantierbarer Kardioverter-Defibrillator
Wenn möglich, wird die Erkrankung behandelt, die die hypertrophe Kardiomyopathie verursacht.
Die Behandlung der hypertrophen Kardiomyopathie zielt in erster Linie darauf ab, das Unvermögen des Herzens zu verringern, sich zwischen den Schlägen mit Blut zu füllen.
Arzneimittel zur Behandlung einer hypertrophen Kardiomyopathie
Betablocker und der Kalziumkanalblocker Verapamil (einzeln oder in Kombination) sind die Basis der Behandlung. Beide verringern die Kraft, mit der sich der Herzmuskel zusammenzieht. Dadurch kann sich das Herz besser mit Blut füllen, und falls der verdickte Muskel den Blutfluss blockiert hat, kann das Blut nun leichter aus dem Herzen abfließen. Zusätzlich verlangsamen Betablocker und Verapamil die Herzfrequenz; das Herz gewinnt also mehr Zeit, sich zu füllen. Manchmal wird auch Disopyramid verordnet, ein Arzneimittel, das die Stärke der Herzkontraktionen mindert.
Amiodaron wird manchmal zur Behandlung von Herzrhythmusstörungen angewendet.
Mavacamten scheint die Symptome zu lindern und die Belastungsfähigkeit zu steigern.
Myektomie
Die operative Entfernung von Schichten des verdickten Herzmuskels (Myektomie) kann den Blutfluss aus dem Herzen verbessern, jedoch wird dieser Eingriff nur vorgenommen, wenn die Symptome den Patienten trotz medikamentöser Behandlung stark behindern. Eine Myektomie kann zwar die Symptome lindern, verringert aber nicht das Todesrisiko. Wenn eine Myektomie in Kliniken durchgeführt wird, die umfangreiche Erfahrung damit haben, sind die langfristigen Ergebnisse hervorragend.
Alkoholablation
Eine Alkoholablation (kontrollierte Verödung eines kleinen Teils des Herzmuskels durch Injektion von Alkohol) ist ein zunehmend genutztes Verfahren, um den Blutfluss aus dem Herzen zu verbessern, weil dieses während einer Herzkatheterisierung möglich ist und keine Operation erfordert. Obgleich es sich bei der Herzkatheterisierung um ein invasives Verfahren handelt, bei dem ein Katheter in das Herz eingeführt wird, ist dieses weniger risikoreich bei Patienten, bei denen ein hohes Komplikationsrisiko bei einer Herzoperation vorliegt.
Implantierbarer Kardioverter-Defibrillator
Für Personen mit einer starken Verdickung des Herzmuskels, insbesondere einer Verdickung der die Herzkammern trennenden Wand (Herzscheidewand), besteht ein hohes Risiko, an einer tödlichen Herzrhythmusstörung zu erkranken, die plötzlich zum Tod führen kann. Bei diesen Menschen empfiehlt sich ein implantierbarer Kardioverter-Defibrillator.
Weitere Informationen
Die folgenden Quellen in englischer Sprache können nützlich sein. Bitte beachten Sie, dass das MANUAL nicht für den Inhalt dieser Quelle verantwortlich ist.
American Heart Association: hypertrophe Kardiomyopathie: Bietet umfassende Informationen zu Symptomen, zur Diagnose und zur Behandlung der hypertrophen Kardiomyopathie.